Initially Posted October,
23, 1996
Revised October 27, 2000

Phenotypic Variation in Sickle Cell Disease: An Analysis
by Errol L. Fields
Undergraduate Student, Harvard University
[this treatise was completed in partial fulfillment of the requirements
of Biology 91R]
Overview
Everyone with sickle cell disease shares the same gene mutation.
A thymine replaces an adenine in the DNA encoding the ß-globin gene.
Consequently, the amino acid valine replaces glutamic acid at the sixth
position in the ß-globin protein product (Ingram, 1956). The change
produces a phenotypically recessive characteristic. Most commonly sickle
cell disease reflects the inheritance of two mutant alleles, one from each
parent.
The final product of this mutation, hemoglobin
S (HbS), is a protein whose quaternary structure is a tetramer made
up of two normal alpha-polypeptide chains and two aberrant ßs-polypeptide
chains. The primary pathological process leading ultimately to sickle shaped
red blood cells involves this molecule. After deoxygenation of hemoglobin
S molecules, long helical polymers of HbS form through hydrophobic interactions
between the ßs-6 valine of one tetramer and the ß-85
phenylalanine and ß-88 leucine of an adjacent tetramer (Schechter,
1978).
Deformed, sickled red cells can occlude the microvascular circulation,
producing vascular damage, organ infarcts, painful crises and other such
symptoms associated with sickle cell disease.
Although everyone with sickle cell disease shares a specific, invariant
genotypic mutation, the clinical variability in the pattern and severity
of disease manifestations is astounding. In other genetic disorders such
as cystic fibrosis, phenotypic variability between patients can be traced
genotypic variability (Powars and Hiti, 1993). Such is not the case, however,
with sickle cell disease. Physicians and researchers have sought explanations
of the variability associated with the clinical expression of this disease.
The most likely causes of this inconstancy are disease modifying factors.
I have reviewed the role of some of these factors, and tried to ascertain
the clinical importance of each.
Fetal Hemoglobin
Augmented post-natal expression of fetal
hemoglobin (HbF) is perhaps the most widely recognized modulator of
sickle cell disease severity. Fetal hemoglobin, as its name implies is
the primary hemoglobin present in the fetus from mid to late gestation
(Higgs et al, 1989). The protein is composed of two alpha-subunits and
two gamma-subunits. The gamma-subunit is a protein product of the ß-gene
cluster. Duplicate genes duplicate upstream of the ß-globin gene
encode fetal globin (Higgs et al, 1989).
Fetal hemoglobin binds oxygen more tightly than does adult hemoglobin
A. The characteristic allows the developing fetus to extract oxygen from
the mother's blood supply (Powars and Hiti, 1993). After birth, this trait
is no longer necessary and the production of the gamma-subunit decreases
as the production of the ß-globin subunit increases. The ß-globin
subunit replaces the gamma-globin subunit in the hemoglobin tetramer so
that eventually adult hemoglobin, HbA, replaces fetal hemoglobin as the
primary component red cells.
HbF levels stabilize during the first year of life at less than
1% of the total hemoglobin. In cases of hereditary persistence of fetal
hemoglobin, that percentage is much higher. This persistence substantially
ameliorates sickle cell disease severity (Personal Communication, Dr. Ken
Bridges, April 1996).
Mechanism of Protection
Two properties of fetal hemoglobin help moderate the severity of
sickle cell disease. First, HbF molecules do not participate in the polymerization
that occurs between molecules of deoxyHbS (Goldberg, et al., 1978).
The gamma-chain lacks the valine at the sixth residue to interact hydrophobically
with HbS molecules. HbF has other sequence differences from HbS that impede
polymerization of deoxyHbS. Second, higher concentrations of HbF in a cell
infer lower concentrations of HbS (Bailey et al, 1991). Polymer
formation depends exponentially on the concentration of deoxyHbS (Eaton
and Hofrichter, 1995). Each of these effects reduces the number of irreversibly
sickle cells (ISC).
Hemoglobin F Levels and Amelioration of Sickle Cell Disease
The level of HbF needed to benefit people with sickle cell disease
is a key question to which different studies supply varying answers. Bailey
et
al. (1991) examined the correlation between early manifestation of
sickle cell disease and fetal hemoglobin level in Jamaicans. They concluded
that moderate to high levels of fetal hemoglobin (5.4-9.7% to 39.8%) reduced
the risk for early onset of dactylics, painful crises, acute chest syndrome,
and acute splenic sequestration.
Platt et al. (1994) examined predictive factors for life
expectancy and risk factors for early death (among Black Americans). In
their study, a high level of fetal hemoglobin (>8.6%) augured improved
survival. Koshy et al. (1989) reported that fetal hemoglobin levels
above 10% were associated with fewer chronic leg ulcers in American children
with sickle cell disease.
Other studies, however, suggest that protection from the ravages
of sickle cell diesease occurs only with higher levels of HbF. In a comparison
of data from Saudi Arabs and information from Jamaicans and Black Americans,
Perrine et al. (1978) found that serious complications (i.e. jaundice,
splenectomy, hematuria) occurred only 6% to 25% as frequently in Saudi
Arabs as North American Blacks. In addition mortality under the age of
15 was 10% as great among Saudi Arabs. Further, these patients experienced
no leg ulcers, reticulocyte counts were lower and hemoglobin levels were
higher on average.
The average a fetal hemoglobin level in the Saudi patients ranged
between 22-26.8%. This is more than twice that reported in studies mentioned
above. Kar et al. (1986) compared patients from Orissa State, India
to Jamaican patients with sickle cell. These patients also had a more benign
course when compared with Jamaican patients. The reported protective level
of fetal hemoglobin in this study was on average 16.64%, with a range of
4.6% to 31.5%. Interestingly, ß-globin locus haplotype analysis shows
that the Saudi HbS gene and that in India have a common origin (see
below).
These studies suggest that the level of fetal hemoglobin that
protects against the complications of sickle cell disease depends strongly
on the population group in question. Among North American blacks, fetal
hemoglobin levels in the 10% range ameliorate disease severity. The higher
average level of fetal hemoglobin could contribute to the generally less
severe disease in Indians and Arabs.
Fetal hemoglobin levels have correlated with specific clinical
complications of sickle cell disease in several studies. Emond et al.
(1980)
examined fetal hemoglobin levels, priapism and consequent impotence.
Priapism
is a prolonged painful erection. The problem develops when sickled cells
obstruct the drainage of blood from the corpora cavernosa.
Emond et al (1980) characterized two types of priapism.
Stuttering episodes of priapism occured in 59% of patients studied while
major episodes affected 38%. Stuttering priapism was characterized by multiple
attacks of less than three hours duration occuring in bursts two to three
times per week for several months. Major episodes were manifested by a
single severe attack exceeding 24 hours duration and most often requiring
hospitalization. In 10% of the patients, major episodes of priapism produced
complete impotence.
Of the patients afflicted by stuttering priapism, 28% subsequently
suffered a major attack. The investigators found that patients with less
severe stuttering attack also had higher levels of fetal hemoglobin than
did those who suffered major attacks. While this study provides some information
useful for treatment and prevention, important pieces of information are
absent.
Although a table provided hematological indices for the overall
patient population, specific information on the group with priapism was
lacking. While the sample size of 61 young men who suffered from attacks
of priapism was reasonable, the study was based on a questionnaire. All
data are retrospective. As with all retrospective analyzes, the completeness
of the information is unknown. Also, no way exists to ascertain whether
observational bias occurred in the initial recording of the data.
The results of Koshy et al (1989), on the other hand can
be accepted greater assurance. Data on patients were collected prospectively.
Consequently, patients were observed under study conditions (e.g.
patients were at steady-state at the time of blood collection; patients
were regularly monitored). Unfortunately, this study failed to include
patients with sickle cell disease who had not suffered from leg ulcers.
Several studies showed little or no correlation between fetal
hemoglobin levels and certain aspects of severity. Seltzer et al.
(1992) reported on five black families with abnormally high levels of fetal
hemoglobin (19-45% HbF). Of the eight patients observed in this study,
two suffered from moderately severe disease. These two patients had HbF
levels of 25% and 31%. Two other patients had HbF levels of 19%. One of
these patients had mild disease while the other suffered from severe symptoms.
The investigators attributed the variability to uneven distribution
of fetal hemoglobin in erythrocytes (mature red blood cells). Other observations
generally supported this line of reasoning. Patients who were asymptomatic
or virtually asymptomatic patients had HbF in most of their erythrocytes.
In contrast, the patients with markedly uneven distributions of HbF tended
to be more symptomatic.
The mean level of fetal hemoglobin in the circulation is important.
However, the distribution of fetal hemoglobin between the cells is also
significant. Heterogeneity of HbF distribution means that some cells will
have none of the protective fetal hemoglobin. These cells would be prone
to sickling, and could occlude the microcirculation, blocking the flow
of cells that normally might have made it across the circulatory narrows.
Such a "logjam" would nullify the anti-sickling effect of HbF in the other
red cells.
However, even accounting for heterogeneous HbF distribution,
not all of the clinical heteroneniety could be explained. For instance,
one moderately symptomatic patient had HbF value of 25% and a F cell percentage
of 79% (namely, 79% of his red cells contained some fetal hemoglobin).
Another patient with mild symptoms had a fetal hemoglobin level of 19%
and an F cell percentage of 17%. More than 80% of the patient's cells lacked
fetal hemoglobin, despite a high mean fetal hemogloin level. Seltzer et
al. concluded that F-cell percentages and fetal hemoglobin levels,
while important, are not the only variables that affect disease severity.
Acquaye et al. (1984) studied two groups of patients in
western Saudi Arabia totaling seventy-one individuals. One group had HbF
levels above 10% and was designated SSHF. The other group had levels of
HbF below 10%, and was designated SSLF. Many patients of both groups suffered
clinically severe sickle cell disease, including urinary and respiratory
tract infections, bone pain or infarcts and severe anemia. Some even had
rare complications such as retinal hemorrhages, epistaxis, hepatic crisis,
acute chest syndrome, and thrombotic stroke.
No significant difference existed in several "severity factors"
in these two groups. These factors included, hemoglobin levels, red cell
count, mean cell volume, mean cell hemoglobin, reticulocyte count, and
serum bilirubin levels. The only clinically significant difference between
the two groups was a higher tendency toward infections and more frequent
hepatomegaly in SSLF patients. Like Seltzer et al., these investigators
concluded that additional factors to fetal hemoglobin levels modulate the
severity of sickle cell disease.
Another study that suggests only a small role at best for fetal
hemoglobin as a modifier of sickle cell disease severity was reported by
El-Hazmi (1992). The subjects were Saudi Arabs in whom a variety of symptoms
associated with sickle cell disease were assessed to form a "severity"
index. The author concluded that among his patients , no correlation existed
between HbF and the severity index.
However, his analysis has a fundamental flaw. El-Hazmi failed
to examine the effect of HbF on each of these symptoms individually. There
important information and an association between fetal hemoglobin levels
specific disease manifestations could be concealed in his data. However,
the study reinforces the conclusion that fetal hemoglobin levels most likely
work in conjunction with other moderating factors to determine clinical
severity in patients with sickle cell disease.
Alpha-Thalassemia
Concurrent alpha-thalassemia has also
been examined as a modifier of sickle cell disease severity. Alpha-thalassemia,
like sickle cell disease, is a genetically inherited condition. The loss
of one or more of the four genes encoding the alpha globin chain (two each
on chromosome 16) produces alpha-thalassemia. A gene deletion most commonly
is at fault. The deletion results from unequal crossover between adjacent
alpha-globin genes during the prophase I of meiosis I. Such a crossover
leaves one gamete with one alpha-gene and the other gamete with three alpha
genes (Bunn, 1986). Upon fertilization the zygote can have 2, 3, 4, or
5 alpha genes depending on the make up of the other parental gamete. In
people of African descent, the most common haploid gamete of this type
is alpha-thal-2 in which there is one deletion on each of the number 16
chromosomes in the patient (Bunn, 1986). Heterozygotes for this allele,
therefore, have three alpha genes (one alpha gene on one of the number
16 chromosomes, two alpha genes on the other).
Embury et al. (1984) examined the effect of concurrent
alpha-thalassemia and sickle cell disease. Based on prior studies, they
proposed that alpha-thalassemia reduces intraerythrocyte HbS concentration,
with a consequent reduction in polymerization of deoxyHbS and hemolysis.
They investigated the effect of alpha gene number on properties of sickle
erythrocytes important to the hemolytic and rheological consequences of
sickle cell disease. Specifically they looked for correlations between
the alpha gene number and irreversibly sickled cells, the fraction of red
cells with a high hemoglobin concentration (dense cells), and red cells
with reduced deformabilty.
The investigators found a direct correlation between the number
of alpha-globin genes and each of these indices. A primary effect of alpha-thalassemia
was reduction in the fraction of red blood cells (RBCs) that attained a
high hemoglobin concentration. These dense cells result from potassium
loss due to acquired membrane leaks. The overall deformability of dense
RBCs is substantially lower than normal.
This property of alpha-thalassemia was confirmed by comparison
of red cells in people with or without 2-gene deletion alpha-thalassemia
(and no sickle cell genes). The cells in the nonthalassemic individuals
were more dense than those from people with 2-gene deletion alpha-thalassemia.
The difference in median red cell density produced by alpha-thalassemia
was much greater in patients sickle cell disease.
Reduction in overall hemoglobin concentration due to absent alpha
genes is not the only mechanism by which alpha-thalassemia reduces the
formation of dense and irreversibly sickled cells. In reviewing the available
literature, Embry and Steinburg (1986) suggested that alpha-thalassemia
moderates red cell damage by increasing cell membrane redundancy (morphologically
seen as target cells). This protect against sickling-induced stretching
of the cell membrane. Potassium leakage and cell dehydration would thereby
be minimized.
These two papers by Embury et al. give some insight into
the moderation of sickle cell disease severity by alpha thalassemia. Some
deficiencies exist, nonetheless. The first paper makes no mention of the
patient pool. Unspecified are the number of patients used, their ethnicity,
or their state of health when blood samples were taken.This information
would help establish the statistical reliablility of the data, and its
applicability across patient groups. Despite these limitation, the work
provides important insight into the mechanisms by which alpha-thalassemia
ameliorates sickle cell disease severity.
Ballas et al. (1988) reached different conclusions regarding
alpha thalassemia and sickle cell disease than did Embury et al
. They reported that decreased red blood cell deformability was associated
with reduced clinical severity of sickle cell disease. Patients with more
highly deformabile red cells had more frequent crises. They also found
that fewer dense cells and irreversible sickle cells correlated inversely
with the severity of painful crises. Like Embury et al., Ballas
and colleagues found alpha thalassemia was associated with fewer dense
red cells.
In addition, Ballas' group found that alpha thalassemia was associated
with less severe hemolysis. However they reached no clear conclusion concerning
alpha gene number and deformability of RBC except to note that the alpha
thalassemia was associated with less red cell dehydration.
The two studies are not completely at odds. Both state that concurrent
alpha-thalassemia reduces hemolytic anemia. They agree that this occurs
through reduction in the number of dense cells, a number directly related
to the fraction of irreversibly sickled cells. Embury et al. conclude
that through this mechanism red blood cell deformability is increased.
The investigators diverge, however, on the relationship to clinical
severity of dense cells and rigid cells. Ballas et al. assert that
both the reduction of dense cells and rigid cells contribute to disease
severity. They advance three possible mechanisms. The most interesting
holds that the higher the deformability of cells, the greater their adherence
to the endothelium lining the blood vessels. Red cell adhesion to endothelial
cells is believed to contribute to vaso-occlusion by retarding erythrocytes
in the microcirculation sufficiently long for sickling to occur there (Fabry,
et
al., 1992). Rigid erythrocytes may or may not enter microvasculature.
If they do they are less likely to adhere to the endothelium and cause
vaso-occlusion or compromise the blood flow. In contrast, deformable cells
have a higher probability of entering the microvasculature, adhering to
endothelium and causing vaso-occlusion.
Ballas (1991) described a pair of patient groups with sickle cell
disease in which the fetal hemoglobin level was less than 15%. The first
group had a significantly fewer painful crises and more leg ulcers. However,
the mortality in this group was 0%, while the second group had 33% mortality
rate. The red cells of the first group were more rigid, and 22% were dense
cells. The second group had fewer rigid red cells and about 10% dense cells.
The reports of Ballas et al. (1988) and Emburyet al.
(1982, 1984 and 1986) agree that concurrent alpha-thalassemia and sickle
cell disease produces less severe hemolytic anemia through the action of
alpha gene number on HbS concentration, HbS polymer formation, and the
frequency of dense cells and ISC. The effect of alpha-thalassemia on other
manifestations of sickle cell disease such as painful crises and vaso-occlusion
are unresolved.
Hemoglobin Haplotype
The final potential modulator of sickle cell disease now known is
haplotype. Of the three modulating factors discussed in this review, the
role of sickle cell haplotype is least well characterized.
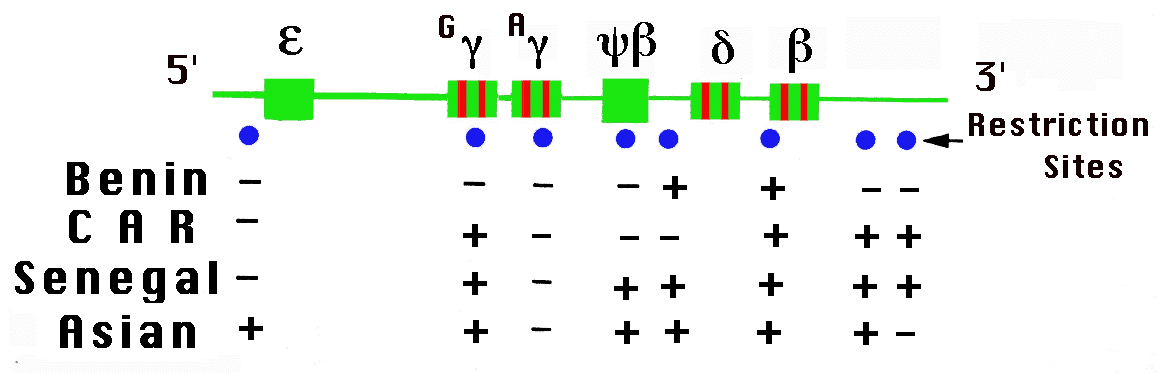 |
| Figure 1. Restriction Endonuclease Sites in the ß-globin Gene
Locus. The ß-globin gene locus or cluster has in series the "ß-like"
genes expressed in humans, including those expressed in embryonic (episilon)
and fetal (gamma) development. After birth, two "ß-like" genes predominate:
beta (98%; gene product, hemoglobin A) and delta (about 2%; gene product
hemoglobin A2). The blue dots indicate the locations of the
informative restriction endonuclease sites used in sickle haplotype analysis
(the specific restriction enzymes used at each site are not indicated).
The pattern of restriction sites for the sickle haplotypes is shown (a
"+ " indicates susceptible to restriction enzyme digestion, while a "-
" indicates resistance to restriction endonuclease digestion.) |
Haplotypes of sickle cell disease can be described as polymorphic
restriction endonuclease sites in and around the mutant ß-globin
gene (Figure 1 and Powars and Hiti, 1993). Although the haplotypes have
numeric identifiers, they are most commonly designated by the geographic
areas in which they were first identified (Figures 2 and 3): Senegal, Benin,
Central African Republic (or Bantu), Cameroon and Arabo-Indian (or Asian)
(Oner,
et al., 1992). The haplotypes were identified by investigators
working with groups of people in various countries primarily in west Afria
(Adekile, et al., 1992). By focusing on people in limited geographic areas,
the investigators were able to limit the confusion which would have resulted
from investigations in areas with a heterogenous population of patients
with sickle cell disease (e.g., the US.)
The Senegal haplotype is represented most prominently in Senegal
and in the most westerly regions of Africa above the Niger River. The Benin
haplotype designates those found in Nigeria, Benin, and countries in the
Bight of Benin. The Bantu or CAR haplotype encompasses those haplotypes
discovered in the Central African Republic and countries in south central
Africa. The Cameroon haplotype has been found in only one ethnic group
in the Cameroons. The Arabo-Indian haplotype usually refers to those haplotypes
found in the Persian gulf and India. Sickle cell disease in India
has been poorly investigated, relative to that in west Africa and the Middle
East. Sickle cell disease is quite prevalent among tribal peoples in India.
Unfortunately, the tribal peoples continue to have limited access to health
care, in part due to their largely rural location.
The existence of haplotypes specific to certain regions of the
world suggests that the mutant ß-globin gene arose separately in
these locations (Oner et al., 1992). All of the areas in question
are now or have been endemic loactions of malarial infestation. This observation
is consistent with the idea that the high incidence of the sickle mutation
in these areas derived from natural selection (Carlson,
et al.,1994).
The mutation that produces sickle hemoglobin occurs spontaneously at a
low rate. People with one sickle hemoglobin gene and one normal hemoglobin
gene (sickle cell trait) are somewhat more resistant to malaria than people
with two normal hemoglobin genes. People with sickle cell trait would have
a better chance of surviving an outbreak of malaria and passing their genes
(for sickle and normal hemoglobin) to the next generation when they have
children.
The independent origin of the sickle mutation opens the possibility
that haplotypes could differ in associated sickle cell disease severity.
The three most common haplotypes in the Americas are Senegal, Benin, and
Bantu/CAR (Hattori et al., 1989)(Powars et al. 1994). In
African populations, each is associated with different degrees of disease
severity. People with the Senegal haplotype,
on average, have the
least severe clinical course, while those with the CAR/Bantu haplotype,
on
average, have the most severe disease. People with the Benin haplotype
usually have disease of intermediate severity (Powars et al , 1994).
Sickle cell disease in India
and the Persian Gulf region apparently follows a more benign course than
it does in Africa and the Americas. The cause of the discrepancy in clinical
manifestations is not clear. Leg ulcers are uncommon in India, for instance.
In contrast, this complication is frequent in Africa and the Americas (Koshy,
et
al., Blood 1989). Priapism, which is a debilitating problem for many
patients in the Americas, likewise is uncommon in India. In contrast, splenomegaly
is common in patients with sickle cell disease in India, while the spleens
of most patients in the Americas are small and poorly functional (due to
recurrent splenic infarctions). Endemic malaria in much of the Indian subcontinent
may account for the splenomegaly. This view is mere supposition, however.
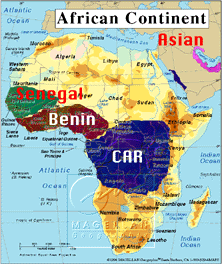 |
| Figure 2. Sickle Hemoglobin Haplotype Distribution in Africa. The
three major ßs-globin haplotypes found in Africa are shown.
The distributions represent the highest concentrations. The genes are expressed
at lower frequency outside the highlighted zones. |
Unfortunately, the dearth of data on sickle cell disease in India
allows nothing more than educated guesses. Although the mutation is identical
in the sixth position of the ßs-globin in both the African
and Asian varieties of the sickle cell disease, the surrounding genetic
environment of the two probably differ. The expression of a gene not currently
recognized as a modifier of sickle cell disease expression, for instance,
could differ between the African and Asian varieties of the condition.
If we knew more about these "epigenetic" factors, new treatment strategies
for severe sickle cell disease might become apparent.
The existence of identical haplotypes in India and in the Persian
Gulf region lacks an obvious explanation. Sickle cell disease in India
exists mainly in the tribal populations,
who to this day remain relatively isolated from the country's mainstream
society. The likelihood is low that an influx of a sickle cell gene from
outside India occurred to a degree to account for rates of heterozygosity
that reach 35% in some tribes. Although current information precludes a
conclusive answer, gene flow from from India to the Persia Gulf area through
commerce and migration seems the more likely scenario. Interestingly, there
are small pockets of sickle genes of the African haplotypes in regions
along India's western coast. Sickle cell disease here exists in the decendants
of African peoples who came to India during the Moghul period, often as
"praetorian" guards for Indian princes.
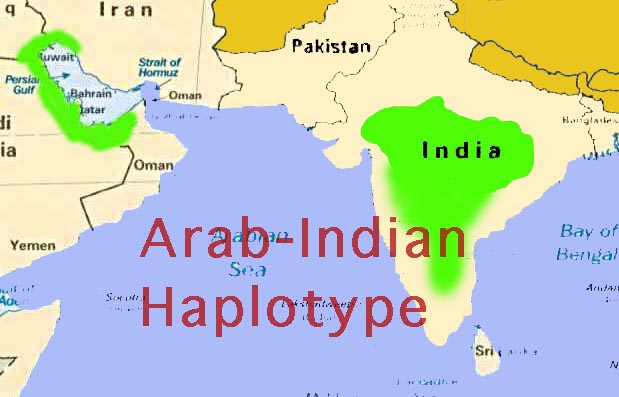 |
| Figure 3. Sickle Hemoglobin Haplotype Distribution in the Middle
East and India. The ßs-globin haplotype found in the
Middle East and India are shown. The haplotypes are identical in the two
areas. The gene probably originated in India and was carried to the Persian
Gulf area by trade and migration. This point is unproven, however. |
The mechanism by which haplotypes influence sickle cell disease
severity remains a mystery. Individual haplotypes have varying levels of
fetal hemoglobin. Patients with the Senegal haplotype often preserve fetal
hemoglobin levels of 20% or more. In contrast, patients with the Bantu/Car
haplotype generally express the lowest fetal hemoglobin levels. The Benin
haplotype is associated with intermediate fetal hemoglobin levels (Powars
and Hiti, 1994).
Powars and Hiti (1994) content that a mutation in the 5' promoter
region of the fetal globin gene, "G-gamma",
maintains the high production of fetal hemoglobin in people with the Senegal
haplotype. Economou et al. (1991) earlier reported that variation
in hemoglobin F levels was not due to nucleotide substitution in
the promoter region of either the "G-gamma" or "A-gamma" fetal globin genes.
Irrespective of the cause, the observation remains higher levels of fetal
hemoglobin are seen in patients with the Senegal haplotype
Most people native to an area indigenous for a particular haplotype
are homozygous for that haplotype. In the Americas, mixing among slave
populations left most patients with sickle cell disease heterozygous for
the two of the three common haplotypes (Nagel et al, 1991) .
Several investigators have examined the effect of heterozygous
sickle haplotype on clinical severity. Nagel et al. (1991) found
that the presence of one Senegal haplotype still results in high fetal
hemoglobin levels, higher overall hemoglobin levels, lower reticulocyte
counts and lower bilirubin levels. In general, patients with at least one
Senegal haplotype had milder disease than those who had none.
In a study conducted by Steinburg et al. (1995), Benin/CAR
heterozygotes trended toward lower fetal hemoglobin levels as well as greater
disease severity, while Senegal/CAR haplotypes tended toward intermediate
characteristics with respect to fetal hemoglobin level and disease severity
(this observation was made by Nagel et al. as well).
In addition to haplotype, Steinberg et al. examined the
effect of at gender in patients with sickle cell disease. Females tended
to have higher levels of fetal hemoglobin than did males, irrespective
of haplotype. The investigators suggested that the higher level of fetal
hemoglobin could reflect hormonal factors that interact with a haplotype-specific
DNA gene regulatory region. Another possibility is a relative peristence
of HbF related to genes located on the X chromosome. Further insight into
this phenomena possibly could be gained by examining post-menopausal patients
with sickle cell disease in whom hormonal patterns differ from those of
younger women.
Although the mechanism by which haplotype is coupled to disease
severity is unknown, a correlation clearly exists. Fetal hemoglobin levels
vary generally by haplotype. A correlation appears also to exist between
between gender, haplotype and HbF levels. This is a relatively new area
of investigation with respect to the variability in sickle cell disease
and has not been as fully characterized as alpha-thalassemia and fetal
hemoglobin effects. Further investigation could shed additional light on
the interplay of haplotypes and disease severity.
Conclusions
Understanding the parameters that modify sickle cell disease severity
would be enormously useful in treatment decisions. For example,hydroxyurea
is a pharmacological agent that raises fetal hemoglobin levels and is used
to treat some patients with sickle cell disease. While no adverse effect
has been noted in short-term studies (Charache, et. al., 1995), the possibility
of long-term complications remains. A method to identify patients with
prognoses for severe disease would allow targeted use of the drug for people
who stand most to benefit.
The need for disease severity prognosis factors is even greater
when
bone marrow transplantation is considered as
a treatment. Bone marrow transplantation can cure patients with sickle
cell disease (Walters
et al., 1996). Transplantation, however, carries
significant intrinsic risks, including death. Reliable selection criteria
for patients most likely to benefit would make transplantation a more attractive
treatment alternative for many patients.
Fetal hemoglobin level is constant throughout life (after stabilization
during infancy) and is a relatively good predictor of disease severity.
Its ability to augur future clinical course is insufficiently fine, however,
for use as the sole arbiter for high risk treatments such as bone marrow
transplantation.
Concurrent alpha thalassemia also provides some prognostic information
about sickle cell disease severity. Alpha thalassemia is a less powerful
predictor than is hemoglobin F level, however. Treatment decisions for
individuals cannot be made on the basis of this parameter.
Haplotypes provide useful population data. Haplotype analysis
has been used by anthropologists to trace the migration of African sickle
cell genes into the Mediterranean basin. As a predictor of disease severity,
however, haplotype analysis is far tos crude for clinical utility.
Multivariant analysis has been used to enhance the predictive
value of independent parameters of disease. By combining several weak predictors,
a more compelling set of forecast data often can be generated. The interrelationship
between fetal hemoglobin levels and haplotype means that these may not
be independent variables in considerations of disease severity. Therefore,
multivariant analysis may be of limited utility.
With rare exception, clinical prediction in medicine is at best
a chancy matter. In sickle cell disease, the point was made dramatically
by Amin et al (1991) who examined many parameters in a monozygotic twin
pair living in the same environment. The sickle cell disease severity was
strikingly different in the children. No measured parameter differed between
the two, however. We must continue to explore the biological basis of sickle
cell disease and its severity. We must at the same time remember that we
only scratch the surface of nature's complexity.
References-
-
Acquaye, JK; Omer, A; Ganeshaguru, K; Sejeny, SA; Hoffbrand, AV. 1985.
Non-benign sickle cell anaemia in western Saudi Arabia. Br. J. Haematol.
60: 99-108.
-
Adekile AD, Kitundu MN, Gu LH, Lanclos KD, Adeodu OO, Huisman TH. 1992.
Haplotypes in SS patients from Nigeria; characterization of one atypical
beta S haplotype no. 19 (Benin) associated with elevated HB F and high
G gamma levels. Ann Hematol 65:41-45.
-
Amin, et al. 1991. Monozygotic twins with sickle cell anemia and discordant
clinical courses: clinical and laboratory studies. Hemoglobin 15:247-256.
-
Bailey, K; Morris, JS; Thomas, P; Serjeant, GR. 1992. Fetal haemoglobin
and early manifestations of homozygous sickle cell disease. Arch. Dis.
Child. 67:517-20.
-
Ballas, SK, et. al. 1988. Rheologic predictors of the severity of the painful
sickle cell crisis. Blood 72: 1216-1223.
-
Ballas, SK. 1991. Sickle cell anemia with few painful crises is characterized
by decreased red cell deformability and increased number ofdense cells.
Am. J. Hematol. 36: 122-30.
-
Bunn, HF; Forget, BG: Hemoglobin: Molecular, Genetic, and Clinical Aspects.
Philadelphia: WB Saunders, 1986.
-
Carlson J, Nash GB, Gabutti V, al-Yaman F, Wahlgren M. 1994. Natural protection
against severe Plasmodium falciparum malaria due to impaired rosette formation.
Blood 84:3909-3814.
-
Charache, et. al. 1995. Effect of hydroxyurea on the frequency of painful
crises in sickle cell anemia. Investigators of the Multicenter Study of
Hydroxyurea in Sickle Cell Anemia. N Engl J Med 332:1317-22.
-
Eaton W., Hofrichter J, 1995. The biophysics of sickle cell hydroxyurea
therapy. Science 268:1142-1143.
-
el-Hazmi, MA. 1992. Heterogeneity and variation of clinical and haematological
expression of haemoglobin S in Saudi Arabs. Acta Haematol. 88: 67-71.
-
Economou, EP; Antonarakis, SE; Kazazian Jr, HH; Serjeant, GR; Dover, GJ.
1991. Variation in hemoglobin F production among normal and sickle cell
adults is not related to nucleotide substitutions in the gamma promoter
regions. Blood 77: 174-177.
-
Embury, SH, et. al. 1982. Concurrent sickle-cell anemia and alpha-thalassemia:
effect on severity of anemia. N. Engl. J. Med. 306: 270-274.
-
Embury, SH, et al. 1984. Concurrent sickle cell anemia and alpha-thalassemia.
Effect on pathological properties of sickle erythrocytes. J. Clin. Invest.
116-123.
-
Emond, AM; Holman, R; Hayes, RJ; Serjeant, GR. 1980. Priapism and impotence
in homozygous sickle cell disease. Arch. Int. Med. 140: 1434-7.
-
Fabry ME, Fine E, Rajanayagam V, Factor SM, Gore J, Sylla M, Nagel RL.
1992. Demonstration of endothelial adhesion of sickle cells in vivo: a
distinct role for deformable sickle cell discocytes. Blood 79: 1602-1611.
-
Goldberg M, Husson M, Bunn H: 1977. The participation of hemoglobins A
and F in the polymerization of sickle hemoglobin. J. Biol. Chemistry 252:3414-3421.
-
Hattori Y, Kutlar F, Kutlar A,McKie, VC, Huisman THJ. 1986. Haplotypes
of ßs chromosomes among patients with sickle cell anemia
from Georgia. Hemoglobin 10: 623-642.
-
Ingram, VM 1956. A specific chemical difference between the globins of
normal human and sickle cell anemia hemoglobin. Nature 178:792.
-
Kar, B; Satapathy, RK; Kulozik, AE; Kulozik, M; Sirr, S; Serjeant, BE;
Serjeant, GR 1986. Sickle cell disease in Orissa State, India. Lancet 2:
1198-201.
-
Koshy, M; Entsuah, R; Koranda, A; Kraus, AP; Johnson, R; Bellvue, R; Flournoy-Gill,
Z; Levy, P. 1989. Leg ulcers in patients with sickle cell disease. Blood
74: 1403-8.
-
Nagel, RL, et. al. 1991. The Senegal DNA haplotype is associated with the
amelioration of anemia in African-American sickle cell anemia patients.
Blood 77: 1371-1375.
-
Oner C, Dimovski AJ, Olivieri NF, Schiliro G, Codrington JF, Fattoum S,
Adekile AD, Oner R, Yuregir, GT, Altay C, et al. 1992. Beta S haplotypes
in various world populations. Hum Genetics 89:99-104
-
Perrine, RP; Pembrey, ME; John, P; Perrine, S; Shoup, F. 1978. Natural
history of sickle cell anemia in Saudi Arabs. A study of 270 subjects.
Annals of Int. Med. 88: 1-6.
-
Platt, OS; Brambilla, DJ; Rosse, WF; Milner, PF; Castro, O; Steinberg,
MH; Klug, PP. 1994. Mortality in sickle cell disease. Life expectancy and
risk factors for early death. N. Engl. J. Med 330: 1639-44.
-
Powars, D, et al. 1993. Sickle cell anemia. Beta s gene cluster haplotypes
as genetic markers for severe disease expression. Am. J. Dis. Child. 147:1197-1202.
-
Powars, D, et. al. 1994. Beta-S gene cluster haplotypes modulate hematologic
and hemorheologic expression in sickle cell anemia. Use in predicting clinical
severity. Am. J. Ped. Hematology-Oncology 16:55-61.
-
Schechter, AN; Bunn HF 1982. What determines severity in sickle cell disease?
(Editorial) N Engl J Med. 306:295.
-
Seltzer, WK; Abshire, TC; Lane, PA; Roloff, JS; Githens, JH. 1992. Molecular
genetic studies in black families with sickle cell anemia and unusually
high levels of fetal hemoglobin. Hemoglobin 16: 363-77.
-
Steinberg, MH; Embury, SH. 1986. Alpha-thalassemia in blacks: genetic and
clinical aspects and interactions with the sickle hemoglobin gene. Blood
68:985.
-
Walters MC, Patience M, Leisenring W, Eckman JR, Scott JP, Mentzer WC,
Davies SC, Ohene-Frempong K, Bernaudin F, Matthews DC, Storb R, Sullivan
KM. 1996. Bone marrow transplantation for sickle cell disease. N Engl J
Med 335: 369-376.




