last revised December 20, 2000
Management of Patients with Sickle Cell Disease
An Overview
Contents
-
Background
-
Nature of the Problem
-
Modulators of SCD Severity
-
Origin of the Sickle Mutation
-
Management of Acute Problems
-
Pain
-
Acute chest syndrome
-
Infection
-
Bone marrow necrosis
-
Stroke
-
Splenic sequestration crisis
-
Aplastic crisis
-
Hepatic sequestration crisis
-
Priapism
-
Management of Chronic Problems
-
Pain
-
Anemia
-
infection prophylaxis
-
Avascular bone necrosis
-
Osteomyelitis
-
skin ulcers
-
Renal dysfunction
-
Retinopathy
-
Heart
-
Pregnancy
-
Newer Therapies
-
Hydroxyurea
-
Erythropoietin
-
Butyrate
-
Clotrimazole
-
Nitric Oxide
-
FluocorTM
-
Bone marrow transplantation
-
Gene replacement therapy
References
Background
Nature of the Problem
 Sickle
cell disease (SCD) results from the substitution of a valine residue for
glutamic acid at position 6 in the beta-subunit of hemoglobin
(Ingram, 1956). With a few minor exceptions, people
with only one gene for hemoglobin S (Hb S) are phenotypically
normal (sickle
trait). People who inherit two Hb S genes from their parents
have sickle cell disease. Deoxygenated Hb S tends to polymerize non-covalently
into long strands that deform the erythrocyte, giving the characteristic
"sickle cell" morphology (Eaton and Hofrichter, 1990). Hb S with bound
oxygen (e.g., in the arterial circulation) does not polymerize.
Sickle
cell disease (SCD) results from the substitution of a valine residue for
glutamic acid at position 6 in the beta-subunit of hemoglobin
(Ingram, 1956). With a few minor exceptions, people
with only one gene for hemoglobin S (Hb S) are phenotypically
normal (sickle
trait). People who inherit two Hb S genes from their parents
have sickle cell disease. Deoxygenated Hb S tends to polymerize non-covalently
into long strands that deform the erythrocyte, giving the characteristic
"sickle cell" morphology (Eaton and Hofrichter, 1990). Hb S with bound
oxygen (e.g., in the arterial circulation) does not polymerize.
 The mechanism by which these changes in the physical properties of the
hemoglobin molecule produce the clinical manifestations of the disease
is not unequivocally proven. The most widely accepted hypothesis is that
erythrocytes deform as they release their oxygen in the capillaries and
are trapped in the microcirculation (Eaton et al., 1976) (Kaul et al.,
1989). The blockade of blood flow produces areas of tissue ischemia, leading
to the myriad of clinical problems seen with sickle cell disease. Although
a good deal of indirect evidence supports this theory, definitive proof
that this is the pathophysiologic mechanism in sickle cell disease is lacking.
The mechanism by which these changes in the physical properties of the
hemoglobin molecule produce the clinical manifestations of the disease
is not unequivocally proven. The most widely accepted hypothesis is that
erythrocytes deform as they release their oxygen in the capillaries and
are trapped in the microcirculation (Eaton et al., 1976) (Kaul et al.,
1989). The blockade of blood flow produces areas of tissue ischemia, leading
to the myriad of clinical problems seen with sickle cell disease. Although
a good deal of indirect evidence supports this theory, definitive proof
that this is the pathophysiologic mechanism in sickle cell disease is lacking.
Recently, investigators have focused on other factors outside the red
cell that could contribute to the manifestations of sickle cell disease.
Hebbel and colleagues first showed that sickle erythrocytes adhere abnormally
to vascular endothelial cells. Their observations were confirmed and extended
by other workers. The endothelial cells may abnormally express adhesion
receptors, perhaps in response to activators released from sickle red cells
(e.g., reactive oxygen species). Other investigators have focused on leucocytes
and platelets which might also contribute to disturbed blood flow in sickle
cell disease. The involvement of multiple components of the blood in the
manifestations of sickle cell disease makes understanding the pathophysiology
more difficult. On the other hand, these additional modulators could be
targeted by new therapies, with diminution in the severity of sickle cell
symptoms.
Sickle cell disease is extremely varied
in its manifestations (Ballas, 1991) (Wethers, 1982). This includes both
the organ systems that are affected as well as the severity of the affliction.
A study of the natural history of sickle cell disease indicated that about
5% of patients account for nearly one-third of hospital admissions (Platt
et al., 1991). A significant number of patients with the disease have few
admissions and live productive and relatively healthy lives. The average
life-span of people with sickle cell disease is shorter than normal, however,
reflecting increased mortality due to the complications of the disease.
Modulators of SCD Severity
Fetal Hemoglobin
Variations in the severity of sickle cell disease between individuals usually
defy explanation. Some factors have been identified that ameliorate the
severity of the condition, however. The most important of these is a high
level of hemoglobin
F (Hb
F) in the erythrocytes (Platt et al., 1991). The first insight into the
role of fetal hemoglobin in the clinical manifestations of SCD was made
by a pediatrician, Janet Watson (Watson, et al., 1948). She and her colleagues
at a New York hospital noted that babies with SCD rarely had manifestations
of the condition in the first year of life. They proposed that the high
level of fetal Hb in the red cells, which persists during the first year
of life, somehow protects the infant. Fetal Hb levels decline to their
routinely low steady-state level between the ages of one to two years.
The childhood manifestations of SCD are seen thereafter.
Patients with sickle cell disease who also have hereditary persistence
of fetal hemoglobin (HPFH) often have few if any symptoms (Stamatoyannopoulos
et al., 1975). In these individuals, Hb F usually comprises greater than
20% of the hemoglobin in the erythrocytes. Patients may be partially protected
from the ravages of sickle cell disease with even lower levels of Hb F.
Unfortunately, few patients with SCD have Hb F levels of greater than 10
or 11% in the absence of HPFH.
Fetal Hb disrupts the polymerization of deoxy-Hb-S (Goldberg et
al., 1977). Since polymerization of deoxy-Hb-S is the signal event in the
pathogenesis of SCD, fetal Hb effectively prevents disease manifestation.
The distribution of Hb F among RBCs is also important. In hereditary persistence
of fetal hemoglobin (HPFH), Hb F exists at high levels in all red cells.
All red cells are equally protected from sickling. In the absence of HPFH,
patients with high levels of Hb F have a heterogeneous distribution of
fetal hemoglobin between cells. An over simplified example is a patient
in whom half the cells have 30% Hb F and half have 0%. The patient would
have 15% Hb F overall. However, half the cells would sickle and occlude
flow through the microcirculation. These deformed cells would block the
flow of the normally shaped high Hb F cells. The patient would experience
all the manifestations of sickle cell disease.
Alpha-thalassemia
Relative to patients with straightforward sickle cell disease, the rate
of hemolysis is lower in people who also have two-gene deletion alpha-thalassemia
(Embury et al., 1982). The mechanism by which alpha-thalassemia ameliorates
red cell destruction is unknown. Polymerization of sickle hemoglobin is
exponentially related to its concentration in the cell. The red cell hemoglobin
concentration in patients with two gene deletion alpha-thalassemia and
sickle cell disease is no different from that of patients with ordinary
sickle cell disease, however (Steinberg and Embury, 1986). The Hb F
concentrations
often is higher in the red cells of these patients and may contribute partially
to the reduction in the rate of hemolysis.
Harbingers of Ill
The advent of therapies that can significantly
ameliorate the clinical course of
sickle cell disease opens the possibility of early intervention. If physicians
could
predict which children will fall victim to recurrent severe pain crises or bouts
of acute chest syndrome, they could intervene before the clinical episodes
thereby
preventing the associated morbidity and possible mortality. This is particularly
relevant for treatments such as bone marrow
transplantation that can cure sickle cell disease
but also carry the risk of significant morbidity and even mortality.
Table 1. Factors that Correlate with a Severe Clinical Course in Sickle
Cell Disease
- An episode of dactylitis prior to one year of age.
- A hemoglobin level of less than 7 g/dl before age 2 years.
- Persistent leucocytosis in the absence of
infection
|
Miller and colleagues (2000) examined the records of nearly 400 children
followed
at comprehensive sickle cell centers. Their multivariate analysis of the
clinical courses of these
children between infancy and 10 years of age uncovered several factors that
augured severe complications, including recurrent severe pain episodes, stroke and
acute
chest syndrome. As seen in Table 1, the variables can be easily identified.
Children
who manifest these characteristics can be considered for aggressive early
treatment
of their sickle cell disease. A smaller study that tracked the course of adult
and
pediatric patients over a 7-year period found that adults
with an elevated white count experienced more frequent hospital admissions for
painful vaso-occlusive
crises than did those with lower white counts (Olatunji, et al., 2000).
Interestingly,
none of the assessed variables correlated with severity in the children. The smaller size of the study and the
the greater age range of the children evaluated likely account for the
difference
from the report by Miller and colleagues (2000). Together, these reports point
to
high white count as a significant risk factor of adverse events in patients with
sickle cell disease.
The relationship of stroke risk to high blood velocity in
the intracranial
arteries is discussed below.

Origin of the Sickle Mutation
Beta-globin haplotypes
The
beta-globin gene exists in a region of chromosome 11 called the "beta globin
locus." Random mutations occur in the non-coding regions of the beta-globin
locus which are neither selected for or against. When a gene mutation occurs
in the coding region of the beta-globin gene (for instance, the conversion
of glutamic acid to valine at postion 6 in sickle hemoglobin), the surrounding
non-coding region is not affected. The genetic background of the surrounding
region is called the "haplotype" of that particular mutation. The chance
is extreme small that another radom mutation will occur in the non-coding
region. Therefore, the haplotype of a particular gene mutation event is
fixed. Analysis of the genomic structure of the beta-globin gene shows
consistent patterns of base substitutions in the non-coding
regions of
the Hb S gene (Bouhassira et al., 1989). The structural regions of the
Hb S genes are identical. The substitutions in the flanking regions of
the gene (the haplotypes) show that Hb S arose separately at least four
times in Africa, and once in Asia, probably in India (Nagel and Fleming,
1992). The four African haplotypes show broad trends in disease severity.
The Central African Republic haplotype tends to have the least favorable
clinical course, followed by the Benin and Senegal haplotypes (Powars and
Hiti, 1993). The ranking of the more recently described fourth haplotype,
Cameroon, is uncertain.
No clear explanation exists for the differences in average severity
between the
haplotypes.
The mutations in the flanking region could secondarily affect severity
by altering Hb F expression in the cells. This is only a hypothesis, however.
The patterns of severity apply only to populations. Broad overlap in the
clinical patterns prevents the use of haplotypes to predict the clinical
course in a particular person. Usually, people with sickle cell disease
outside Africa (e.g., blacks in the United States) or India have mixed
haplotypes for their sickle cell genes. Analysis of haplotype in this setting
is even less likely to provide clinically useful information.
Hb S is common in some areas of the Mediterranean basin, including
regions of Italy, Greece, Albania and Turkey (Boletini et al., 1994) (Schiliro
et al., 1990). Haplotype analysis shows that the Hb S in these areas originated
in Africa. The genes probably moved along ancient trading routes between
wealthy kingdoms in western Africa and the trade centers in the Mediterranean
basin. The high levels of Hb S attained in some areas reflects partial
protection against protection against malaria
provided
by sickle cell trait (see below).
The Hb S mutation arose independently a fifth time in southwest
Asia (Miller et al., 1987). The area of the Middle East near the head of
the Persian Gulf is very marshy. In the past the area was swampy and harbored
malaria. Malaria remains endemic to much of the Indian subcontinent. The
fact that the Hb S mutation apparently arose in response to malaria in
southwest Asia supports the "malaria explanation" of the prevalence of
the gene. The identity of the hemoglobin S haplotype in India and the Persian
Gulf region suggests that it arose in one area and moved to the other with
trade or migration (Ramasamy et al., 1994) (Kar et al., 1986). Although
we cannot be certain of the origin of the Asian haplotype of the sickle
cell gene, the very high prevalence of the gene in tribal peoples of India
suggest that the subcontinent was the place of origin. This Asian variety
of Hb S may on average produce fewer complications than its African counterparts
(Perrine et al., 1978).
The Sickle Gene and Malaria
The high representation of the hemoglobin S gene in some populations
reflects the protection it provides against malaria
(Gendrel et al., 1991)
(Carlson et al., 1994). The malaria parasite does not survive as well in
the erythrocytes of people with sickle trait as it does in the cells of
normals (Orjih et al., 1985). The basis of the toxicity of sickle hemoglobin
for the parasite is unknown. One possibility is that the malarial parasite
produces extreme hypoxia in the red cells of people with sickle trait.
These cells then sickle and are cleared (along with the parasites they
harbor) by the reticuloendotheila system (Roth, et al, 1978). Another possible
mechanism is
that low levels of hemichromes are formed in sickle trait erythrocytes.
Hemichromes are complexes that contain heme moieties that have dissociated
from the hemoglobin. Hemichromes catalize the formation of reactive oxygen
species, such as the hydroxyl radical, which can injury or even kill the
malarial parasites (Anastasi, 1984).
The malaria hypothesis maintains that during prehistory, on average,
people without the sickle gene died of malaria at a high frequency. On
the other hand, people with two genes for sickle hemoglobin died of sickle
cell disease. In contrast, the heterozygotes (sickle trait) were more resistant
to malaria than normals and yet suffered none of the ill-effects of sickle
cell disease. This selection for heterozygotes is termed
"balanced polymorphism".
Support for this concept comes from epidemiological studies in malaria-endemic
regions of Africa. The frequency of sickle cell trait is lower in people
coming for treatment to malaria clinics than is seen in the general population
(Wilcox, et al., 1983).
The reasonable assumption is that relative protection form malaria is at
work in this situation.
Although malaria remains a major health problem in many tropical regions of
the world,
the disease is not a significant threat to people in the temperate zones.
Consequently, the protection afforded by sickle trait no longer has a survival
advantage for many groups of people in whom the sickle cell gene is common.
This has left sickle cell disease as the major health issue in these
populations.
Management of Acute Problems
Pain
Vaso-occlusive pain episodes experienced by patients with sickle cell disease
vary tremendously in frequency and severity. Some patients rarely have
painful crises, while others spend the greater part of a given year in
the hospital receiving analgesics. The cooperative study of the natural
history of sickle cell disease showed that about 5% of patients accounted
for one-third of hospital days devoted to pain control (Platt et al., 1991).
To complicate matters further, the pattern of pain varies over time, so
that a patient who has a particularly severe year may later have a prolonged
period characterized by only minor pain. The frequency and severity of
vaso-occlusive pain episodes often change as a person moves from childhood
to being an adult. The "breakpoint" often occurs during the late teens
or early 20's. Changes in hormonal status that occur during these years
could contribute to the changes in severity of sickle cell disease. However,
no causal relationship has been established, so the association remains
only temporal.
The mode of onset of sickle cell pain crises likewise varies. Patients
can develop agonizingly severe pain in as little as 15 minutes. In other
instances, the pain gradually escalates over hours or even days. Patients
manage most episodes of pain at home. Oral analgesics, combined with rest
and fluids often allows a person to "ride out" the pain episode. Some patients
report that warm baths or warm compresses applied to aching joints ameliorates
the severity of the pain.
The sites affected in acute painful crises vary for each patient. Pain
occurs commonly in the extremities, thorax, abdomen, and back (Ballas and
Delengowski, 1993). Pain tends to recur at the same site for a particular
person. For each person, the quality of the crisis pain is usually similar
from one crisis to another. During the evaluation, the provider should
question the patient as to whether the pain feels like "typical" sickle
cell pain. Most patients can distinguish back pain due to pyelonephritis
or abdominal pain due to cholecystitis, for instance, from their typical
sickle cell pain. If the quality of the pain is not typical of their sickle
cell disease, other causes should be investigated before ascribing it to
vaso-occlusion.
No reliable objective index of pain
exists. The provider depends solely
on the patient's report. One of the most difficult problems that patients
with sickle cell disease face is seeking treatment for pain in a setting
in which they are unknown. The typical scenario is one in which a patient
is brought to a busy emergeny room complaining of pain. Some patients writh
with severe pain while others are stoic. The person who bears the pain
with as much poise as possible runs the risk of not being believed by the
staff. Some providers mistakenly believe that the number of deformed sickle
cells on the peripheral blood smear reflects the degree of pain that a
patient is experiencing. Other providers look to parameters such as blood
pressure and heart rate. Although these measures provide more information
than the peripheral smear, they do not reliably reflect pain severity.
Trust in the patient report is key to the management of sickle cell pain
crises.
Opiods
The pain experienced with an acute painful crisis typically is quite severe.
Most patients describe a full blown crisis as the most intense pain that
they have ever experienced. The pain sometimes increases in severity slowly
over a couple of days. At other times, a crescendo is reached in less than
15 minutes. Pain control often requires large quantities of opiod analgesics.
The exact amount varies, and depends in part on the frequency with which
the person requires opiods. For many patients, 4 to 8 mg of hydromorphone
can be given as an intravenous bolus over 15 to 20 minutes followed by
another 4 mg in 30 minutes if pain control is inadequate.
Patients often feel that one analgesic, such as hydromorphone for example,
controls pain more effectively than others. Therefore, they should be questioned
about the kind of medication that has worked best in the past. Also, some
patients may experience reactions with one analgesic (e.g., itching with
meperidine) but not with others (Pegelow, 1992).
Pain relief occurs more slowly with intramuscular injections, and the
injections themselves can produce substantial discomfort. Consequently,
intravenous administration of analgesics is usually preferable. As pain
control improves, the analgesia should be maintained to prevent the patient
from slipping back into a painful cycle. The "prn" administration of analgesics
should be avoided, if possible (Robieux et al., 1992). Following stabilization
in the emergency situation with intravenous boluses of opiods, the patient
should be transferred to the floor and placed a maintenance regimen.
"Patient-controlled
analgesia" (PCA) often works well for pain relief (Holbrook, 1990) (McPherson
et al., 1990). With these infusion devices, patients can administer small
doses of additional medication over their continuous infusion (to a fixed
maximum) to control flares of pain.
Patients can become drowsy as their pain is controlled. Often, this
reflects the fatigue that comes with one or more sleepless nights with
pain at home. The analgesics should not be discontinued automatically for
somnolence as long as the patient is easily aroused. A common misconception
is that if a person is sleeping, the analgesics are controlling the pain.
Patients often sleep despite severe pain. The quantity of analgesia can
be slowly reduced as the patient's symptoms improve. While the tapering
of intravenous analgesics can require only two or three days, control of
a full blown crisis often requires 10 to 14 days. Less commonly, bouts
of sickle vaso-occlusive pain require several weeks to control.
Meperidine can present problems for
pain control in patients with
sickle cell disease. The half-life of the drug in the circulation is
about 4 hours. The liver converts meperidine to normeperidine, a derivative
that has analgesic activity but which also is toxic. Grand mal seizure
is a particularly serious complication that occurs with the administration
of large amounts of meperidine. Normeperidine likely is the primary culprit
in this situation. Other opiod analgesics, therefore, are preferable to
meperidine. The American Pain Society recommends that meperidine no longer
be used for control of pain in people who require long-term analgesic treatment.
Eventually the patient should be switched to oral opiod analgesics,
which may be necessary for a week or more after discharge (Friedman et
al., 1986). The parenteral analgesics should be tapered after the oral
medication is started. Abrupt termination of parenteral analgesics when
oral medications are begun can cause a rebound in sickle cell crisis pain.
Most patients with sickle cell disease manage their analgesics responsibly.
If possible, they should have a supply of analgesics at home to control
less severe episodes of pain. In addition to analgesia, patients with painful
crises should also receive supplemental oxygen and intravenous fluids.
Once the pain is under control, oral hydration can replace the intravenous
fluids.
Epidural analgesia has been used for pain control in some patients with
sickle cell disease (Tobias, 1993) (Yaster et al., 1994). This approach
is most effective when the major discomfort is below the level of the chest.
Although some patients receive good relief with epidural analgesia alone,
others continue to require systemic analgesics, albeit at lower doses.
Some patients have a psychological aversion to having needles introduced
into their backs and balk at epidural analgesia, despite its superior pain
relief relative to systemic analgesics.
Non-steroidal anti-inflammatory drugs (NSAIDs)
Recently, NSAIDs have been added to the management algorithm of acute sickle
cell pain (Sanders et al., 1992). The drug that has been used most often
in this context is ketorolac tromethamine. The reports of the use of this
agent to control acute painful episodes in patients with sickle cell disease
have been largely anecdotal (Perlin et al., 1994). While some reports are
positive, others show no effect of ketorolac in the treatment of acute
vaso-occlusive pain crises (Wright et al., 1992). Ketorolac comes in a
preparation that is designated for intramuscular injection. However, the
medication can be diluted into normal saline and infused as an intravenous
bolus. Ketorolac alone usually will not control an episode of acute sickle
cell pain. However, the medication appears to operate synergistically with
opiod analgesics. Patients often recover more rapidly and require less
opiods when ketorolac is added to the treatment regimen. A single 30 mg
intravenous bolus is usually administered for supplemental pain control.
Ketorolac can produce gastritis and bleeding. The drug should be used cautiously
in patients with peptic ulcer disease or a history of gastrointestinal
bleeding. NSAIDs can impair kidney
function and accelerate the renal
injury produced by sickle cell disease itself. For these reasons,
many
specialists avoid NSAIDs in patients with sickle cell disease.
Transfusion
Transfusion therapy appears intuitively reasonable for a disorder that
results from polymerization of deoxygenated hemoglobin in the red cells.
The complex pathophysiology of sickle cell disease confounds the picture,
however. Vaso-occlusive sickle cell crises are probably fueled, at least
in part, by sluggish blood flow through the microcirculation (Clark et
al., 1980). Slow blood flow promotes deoxygenation of hemoglobin and
polymerization
of the molecules. Although the oxygen carrying capacity of blood increases
with hematocrit, so does viscosity. As the hematocrit rises beyond the
range of the low-30's, increased viscosity may outweigh enhanced oxygen
delivery and swing the dynamics of the situation toward sickling.
In areas where tissues are poorly perfused due to vaso-occlusion, the
ability of additional red cells to reverse local hemoglobin S polymerization
is questionable. Transfused erythrocytes will not improve blood flow through
regions of the microcirculation that are occluded by deformed red cells.
Although the transfused red cells do not sickle in the microcirculation
even with slow flow, the overwhelming predominant sickle erythrocytes in
the circulation is decisive in the development of local vaso-occlusion.
Simple transfusion is not an effective intervention for the management
of acute painful episodes in patients with sickle cell disease. Exchange
transfusion has been used in attempts to alleviate bouts of severe, intractable
pain with better effect, overall (Davies and Brozovic, 1989). In addition,
chronic transfusion therapy has been used to decrease the frequency of
pain in patients with recurrent debilitating painful crises (Keidan et
al., 1987). While sometimes effective, this approach as a number of problems,
as detailed below.
Corticosteroids
A recent report described the use of corticosteroids in a cohort
of children with severe sickle cell pain crises (Griffin et al., 1994).
The patients received large doses of intravenous steroids on each of the
first two days of their painful sickle crises. The treatment group required
narcotic analgesics for approximately half as long as the control patients.
The rate of pain relapse was significantly higher in patients who received
the steroid treatment. This intriguing observation awaits confirmation,
particularly in adults with sickle cell disease.
Acute Chest Syndrome
Acute chest syndrome (ACS)
is difficult to diagnose because its etiology varies and its manifestations
are variegate. Common characteristics include fever, dyspnea, cough, and
pulmonary infiltrates (Haynes Jr and Kirkpatrick, 1993) (Poncz et al.,
1985). The infiltrates can have a lobar distribution, but often are bilateral.
Sometimes, the pulmonary picture is one of diffuse, hazy opacities that
resemble adult respiratory distress syndrome. In other instances, ACS looks
like a simple pneumonia. This problem in diagnosis is aggravated by the
fact that infectious agents such as viruses, bacteria, and mycoplasma can
trigger the syndrome (Charache et al., 1979) (Kirkpatrick et al., 1991)
(Miller et al., 1991). Bone marrow infarction with secondary pulmonary
fat emboli also can trigger the acute chest syndrome (Vichinsky et al.,
1994) (Gelfand et al., 1993). In most instances, the etiology of ACS is
a mystery.
The arterial blood oxygen saturation commonly falls with ACS to a greater
degree than occurs with a simple pneumonia of the same magnitude. Patients
with the acute chest syndrome often have progressive pulmonary infiltrates
despite treatment with antibiotics (Koren et al., 1990). Infection may
set off a wave of local ischemia that produces focal sickling, deoxygenation
and additional sickling.
The microcirculatory vessels in the lung tend to constrict with
hypoxia rather than dilate, as occurs with vessels in other parts of the
body. Regions of vascular constriction could worsen the occlusion of the
microcirculation. Unchecked, ACS can produce cardiovascular collapse and
death. ACS occurs more commonly in children than adults (Sprinkle et al.,
1986) (Gill et al., 1995). People who survive an episode of ACS are more
likely than the general sickle cell population to have future attacks.
Patients who suffer recurrent episodes of ACS are prone to develop chronic
lung insufficiency.
Given the baseline anemia in SCD, pulmonary compromise is a serious
complication. The most important step in the treatment of ACS is to recognize
the disorder. Potential bacterial infections should be treated with appropriate
antibiotics. When symptoms progress, particularly with a worsening of the
chest roentgenogram, ACS must be considered. Sometimes, the pulmonary pattern
mimics congestive heart failure. However, congestive heart failure is uncommon
in patients with SCD who are in the 15 to 30 year age range, making the
presumptive diagnosis of ACS more likely. A relentless decline in arterial
oxygenation is often a harbinger of ACS, and demands prompt action.
Exchange transfusion is the treatment of choice for ACS (Lanzkowsky
et al., 1978) (Emre et al., 1995). The procedure involves exchange of the
total blood volume and is done most efficiently using an apheresis machine.
When an apheresis machine is not available, sequential transfusion/phlebotomy
can be performed. A hemoglobin electrophoresis should be sent prior to
the exchange transfusion. A second should be sent after the procedure.
The object is to ensure that the exchange has reduced the percentage of
Hb S cells to under 30%. Patients often improve substantially within hours
of an exchange. Rising arterial oxygenation and decreasing dyspnea usually
augur recovery. The chest roentgenogram typically lags behind the clinical
status. Since a bacterial pneumonia rarely can be excluded in these patients,
most receive concomitant broad-spectrum antibiotics.
A serious potential problem with exchange transfusion is delayed transfusion
reaction (Diamond et al., 1980). Most patients with SCD are of African
ancestry. Most of the blood available for transfusion comes from people
of European descent. A number of minor red cell antigens are expressed
at different frequencies in these two groups. Repeated transfusion of any
African-American can, therefore, induce antibodies
directed against these minor antigens.
Should years pass between transfusions, the titers of antibody can fall
to levels that are undetectable by routine cross matching. Transfusion
with blood containing the offending antigen often rekindles antibody production
to high levels in only a few days. In a person who has received exchange
transfusion, a large fraction of the circulating red cells can be destroyed
in a deadly delayed transfusion reaction.
The transfusion records of any exchange transfusion candidate
should be searched thoroughly for any history of antibodies to minor red
cell antigens. Antibody screening should be repeated three to four weeks
after the exchange transfusion to look for new alloantibodies to minor
antigens.
Infection
Patients with sickle cell disease are susceptible to overwhelming infection
(Olopoenia et al., 1990) (Overturf et al., 1977) (Landesman et al., 1982).
The most significant factor is splenic autoinfarction during childhood
(Fernbach and Burdine Jr, 1970). Functional asplenia leaves patients vulnerable
to infections with encapsulated organisms such as Streptococcus
pneumoniae
and
Hemophilus influenzae. Further, some studies suggest that neutrophils
do not function properly in patients with sickle cell disease (Humbert
et al., 1990). How the mutation in sickle cell disease might lead to a
defect in neutrophil function is unclear.
Patients with SCD and unexplained fever should be cultured thoroughly.
If the clinical condition suggests septicemia, the best action is to start
broad spectrum antibiotics after complete culturing. Signs of systemic
infection include fever, shaking chills, lethargy, malaise, and hypotension.
Patients with septicemia can
expire in only a few hours. Therefore,
observation is not a good option when sepsis is suspected.
Acute Bone Marrow Necrosis
Acute bone marrow necrosis is now recognized more often as a complication
of sickle cell disease, in part due to improved methods of detection (Johnson
et al., 1994) (Shapiro and Hayes, 1984). In the past, the diagnosis could
only be made by bone marrow biopsy or inferred from the complications that
resulted. If the necrosis occurred in regions of the marrow that were not
easily biopsied, the diagnosis was almost impossible to confirm. This has
changed with the introduction of magnetic resonance imaging (MRI) techniques
(Mankad et al., 1990) (Rao et al., 1989). Bone marrow should have the density
of other body tissues on MRI scans. With bone marrow necrosis, marrow
liquefaction
is easily detected on scan.
Patients with bone marrow necrosis often suffer
excruciatingly severe pain. Some patients require drastic measures, such
as epidural anesthesia for control of wrenchingly intense pain. Patients often
describe acute bone marrow necrosis as producing "the worst pain I've ever
experienced." The necrosis
frequently occurs in the marrow of the ribs, femur or tibia.
Pulmonary fat emboli can complicate bone marrow necrosis (Johnson et
al., 1994). Fat emboli can trigger respiratory insufficiency or even
acute
chest syndrome. Making the diagnosis of fat emboli to the lungs is
difficult. In some cases, sputum samples stained with Oil Red O will show
fat-laden macrophages. Exchange transfusion has been used with success
in some patients with acute bone marrow necrosis. The experience is anecdotal,
since the ability to document bone marrow necrosis in patients is a relatively
recent development.
Stroke
Strokes are much more common
in children than in adults. The average age of stroke victims is about
4 years (Ohene-Frempong, 1991). Frequently, large arteries such as the
internal carotid or the middle cerebral are occluded (Balkaran et al.,
1992) (Earley, et al., 1998). The mechanism of occlusion of these vessels
is not clear, despite necropsy examination of a number of children who
succumbed to the condition. Imaging procedures such as angiography and
the non-invasive magnetic resonance angiography (MRA) have provided information
on the sequence of events that proceed a stroke (Adams et al., 1992) (DeBaun
et al., 1995) (Wang et al., 1992). Narrowing of arteries near sharp turns
often are seen. A common finding is narrowing at the separation of the
middle cerebral and the internal corotid arteries. Paradoxically, the higher
rate of blood flow produced by arterial narrowing is believed to contribute
to the risk of complete arterial occlusion. A complete occlusion at this
critical location produces massive strokes.
A key question is whether medical intervention can prevent a stroke
in a child with an arterial lesion. The Stroke Prevention Trial in Sickle
Cell Anemia (STOP), sponsored by the NHLBI, was conducted at several
institutions
(Adams, et al., 1998) to address this question.
Between February, 1995 and October, 1996, the trial, coordinated
through the Medical College of Georgia (Dr. Robert Adams) and the New England
Research Institutes (Dr. Donald Brambilla), enrolled 130 subjects, ages
2 to 16, who were at high risk for stroke on the basis of elevated cerebral
blood flow measured by transcranial doppler (TCD) screening tests (greater
than or equal to 200 cm/sec time averaged mean velocities). The patients,
drawn from 13 US clinical centers and one in Canada, were randomized to
receive either standard supportive care or periodic blood transfusions.
The primary endpoint was the rate of stroke rates in the treated and control
groups.
The primary data analysis in the STOP Trial compared stroke rates
in 63 children randomized to receive repeated exchange or simple transfusions
and 67 children who received standard supportive care. A stroke was defined
as clinically significant neurologic impairment and physical findings,
supported by an abnormal magnetic resonance imaging (MRI) study. The clinical
records and MRI's were analyzed by a panel that was blinded to the treatment
assignment of the study subjects.
The patients in the transfusion arm received simple or exchange
transfusions every 3-to- 4 weeks in an effort to maintain the Hb S level
below 30%. After one year, 10 of the children in the standard care group
had suffered a cerebral infarction, compared with one child in the transfusion
group. This difference represents a 90% relative decrease in the stroke
rate in the transfused patients. These results were so compelling that
the study's Data and Safety Monitoring Board, composed of independent,
outside experts in the fields of pediatric hematology, neurology, radiology,
statistics, and ethics recommended that the trial be terminated early so
that the children who had been receiving standard supportive care could
be offered an effective treatment to prevent first-time stroke. On September
2, 1997, the study was halted, and the investigators in the 14 participating
centers were notified of the results and the efficacy of transfusion therapy.
The STOP Trial confirmed that TCD can identify children with sickle
cell anemia at high risk for first-time stroke. Since the greatest risk
of stroke occurs in early childhood, the NHLBI recommends that children
ages 2-16 receive TCD screening. Screening should be conducted at a
site where clinicians have been trained to provide TCDs of comparable quality
and information content to those used in the STOP Trial. The clinicians
should also be able to read them in a manner consistent with what was done
in STOP. To apply the predictive and therapeutic information developed
in the STOP Trial, two abnormal STOP-comparable TCDs are needed to identify
patients at high risk of stroke (velocity greater than 200 cm/sec on two
separate occasions).
During follow-up, some children in the large screening population,
who initially had normal or ambiguous TCD readings developed frankly abnormal
TCD readings. These data suggest that children with normal TCDs should
be re-screened at an interval which depends on their age and the prior
result of TCD. Although the optimal timing is not known, re-screening
approximately
every 6 months is a reasonable objective.
Stroke in SCD is a medical emergency. The deficits are often
profound,
although many children recover substantial function. Exchange transfusion
followed by maintenance hypertransfusion is mandatory (Cohen et al., 1992)
(Pegelow et al., 1995). This action improves recovery and reduces the risk
of recurrent stroke. In the absence of this intervention, as many as two-thirds
of children will suffer subsequent events (Wang et al., 1991). The optimal
duration of therapy is unclear. Several studies have shown that as many
of 50% of children on maintenance therapy for as long as 5 years suffer
new strokes within months of stopping treatment.
A newly recognized area of concern in patients with sickle cell disease
is "silent stroke" (White and DeBaun, 1998). Recent technological advances,
including MRI, MRA, and PET scanning have been combined with neuropsychiatric
testing to gain a new window into the effect of silent strokes in children
with sickle cell disease. Analysis of 42 children followed as part of the
Cooperative Study of Sickle Cell Disease showed that nearly 20% had suffered
silent cerebral infarction, as detected by MRI (Kinney, et al., 1999).
Multivariate analysis showed a number of associations, including a greater
risk of silent infarctions with lower hematocrits. This association was
found in another study of 50 patients (Steen, et al., 1999). One-third
of the latter children showed evidence of mild intellectual impairment
on cognitive testing. Positron emission scans and magnetic resonance angiography
could be useful adjuncts in the effort to diagnose children with silent
cerebral infarction (Powars, et al., 1999), (Gilliams, et al., 1998). Clearly
this is an area of major concern that deserves much more investigation.
In adults, hemorrhagic stokes occur more frequently than arterial
occlusive strokes (Van Hoff et al., 1985). Subarachnoid hemorrhages are
most common. Bleeds that involve deep structures in the brain, such as
the thalamus, also occur, however. In some instances, this reflects the
development of "moya-moya" syndrome years after an earlier thrombotic stroke.
A network of delicate capillaries can form, often in the area of the old
infarction. Angiography reveals the complex filamentous structure of the
moya-moya lesion (Peerless, 1997). Should these capillaries rupture, disastrous
intracranial hemorrhage occurs. Easily accessible lesions are sometimes
surgically excised. Thrombotic strokes in adults are as mysterious as those
in children. Nonetheless, exchange transfusion followed by maintenance
hypertransfusion is a prudent course of action.
Splenic Sequestration Crisis
Splenic sequestration crisis
results from the acute entrapment of large
amounts of blood in the spleen (Sears and Udden, 1985). The manifestations
are left upper quadrant pain, exacerbated anemia and, often, hypotension.
In children, a large fraction of the circulating blood volume is frequently
sequestered. Splenic sequestion crisis is a medical emergency that demands
prompt and appropriate treatment. Parents should be familiar with the signs
and symptoms of splenic sequestion crisis. Children should be seen as speedily
as possible in the emergency room.
Circulatory collapse and death can
occur in less than thirty minutes.
Splenic autoinfarction makes splenic sequestration crisis uncommon in adults
with homozygous Hb S sickle cell disease. The condition can occur in adults
with sickle beta-thalassemia or sickle-hemoglobin C (SC) disease since
autoinfarction does not occur in these syndromes (Roshkow and Sanders,
1990) (Solanki et al., 1986). The most prominent symptom is left upper
quadrant pain. The larger blood volumes of adults make hypotension and
circulatory collapse much less common than in children.
The treatment of splenic sequestration crisis includes intravenous fluids
and transfusion as necessary to maintain the intravascular volume. A child
who suffers one episode of splenic sequestration crisis is at greater risk
of a second attack (Kinney et al., 1990). Specialists debate whether children
who survive an episode of splenic sequestration crisis should undergo
prophylactic
splenectomy after their recovery (Szwed et al., 1980). Less is known about
the condition in adults. Given the lower morbidity and mortality in adults,
splenectomy is rarely a consideration.
Aplastic Crisis
Aplastic crisis is
a potentially deadly complication of sickle cell disease that develops
when erythrocyte production temporarily drops. Infection with parvovirus
B-19 frequently causes aplastic crises (Saarinen et al., 1986). This
adeno-associated
virus causes "Fifth Disease", a normally benign childhood disorder associated
with fever, malaise, and a mild rash. The virus has a trophism for erythroid
progenitor cells, and impairs cell division for a few days during the infection.
Normal people experience, at most, a slight drop in hematocrit since the
half-life of erythrocytes in the circulation is 40 to 60 days. The picture
is different in patients with hemolytic anemias, who maintain reasonable
hematocrits only through prodigious production of new red cells. A shut-down
in erythroid production for a few days in these patients can lead to potentially
deadly declines in hematocrit (Mallouh and Qudah, 1993). Often, but not
always, aplastic crises coincide with a painful crises. The reticulocyte
count should be checked on admission to the emergency room or to the hospital
in patients with SCD. The treatment of aplastic crisis is purely supportive,
with transfusions to maintain an acceptable hematocrit until marrow activity
is restored.
Hepatic Sequestration Crisis
Sickled cells can become lodged in the liver obstructing blood flow through
the organ (Davies and Brozovic, 1989). The result is painful hepatic enlargement
accompanied by an increase in the plasma levels of hepatic synthetic enzymes
(e.g., ALT, AST). The serum bilirubin levels often skyrocket to levels
in the range of 30 to 40 mg/dl. Acute hepatic failure can ensue. Fluids,
oxygen and analgesia are the usual management interventions taken. The
benefit of more aggressive measures such as exchange transfusion is unknown.
Priapism
Priapism is a potentially serious problem for young men with sickle cell
disease. The condition is believed to result from impaired blood egress
from the corpus spongiosum of the penis, leading to prolonged erections
(Fowler Jr et al., 1991). The affliction often occurs in association with
spontaneous nocturnal erections. Episodes of priapism can last from several
hours to several days. One group of investigators reported a ninety percent
actuarial probability of at least one episode of priapism by age twenty-one
years
(Mantadakis, et al., 1999).
Stuttering priapism is common. Here, the (typically) young man develops
erections lasting one to two hours, initially, that resolve spontaneously.
The condition then progresses to a point where the erections are quite
prolonged and painful. Priapism lasting more than three or four hours is
a medical emergency since it can produce impotence (Mykulak and Glassberg,
1990) (Emond et al., 1980).
The most commonly used intervention in the past was irrigation of the
ventral vein of the penis by a urologist in an attempt to remove the blockage
to blood flow. Since the problem is one of microvascular occlusion, the
results of this approach were generally poor. Not only does the surgery
often fail to resolve the priapism, but the procedure itself risks inducing
impotence (Yang et al., 1990). More recently, exchange transfusion has
been used in some of these patients with mixed results (Seeler, 1973).
Non-acute cases of priapism are sometimes treated with conjugated estrogens
(Serjeant et al., 1985) or vasodilators (Baruchel et al., 1993). While
there is some clinical data to support the short-term use of estrogens,
the opinions of specialists in sickle cell disease remain divided.
Management of Chronic Problems
Pain
Chronic pain is a major problem for many patients with sickle cell
disease. The etiology of chronic pain in sickle cell disease is uncertain.
Organ injury and necrosis produced by years of intermittent ischemia from
vaso-occlusion likely plays a large role. Radiographs of bones show
characteristic
deformities of the ribs, for instance. The damage that produced the deformities
likely produces chronic pain. The severity of the pain varies greatly and
can change over time. Some patients control their pain by intermittently
using mild analgesics, such as non-steroidal anti-inflammatory agents.
Others require frequent doses of opiod analgesics. Consequently, no universally
applicable formula exists for management of chronic pain. The patients
who frequently present the greatest management challenges for physicians,
are those with persistent severe pain controlled by chronically administered opiod analgesics.
Non-steroidal anti-inflammatory drugs
Non-steroidal anti-inflammatory drugs (NSAIDs) can control chronic pain
in many patients with sickle cell disease (Sanders et al., 1992). The agents
can be used alone or in conjunction with opiod analgesics. Most commonly,
NSAIDs are used intermittently to control flairs of pain. One potential
problem with these agents is renal damage. Patients with sickle cell disease
are more susceptible than normal people to renal
injury. Since renal damage can be compounded by NSAIDs, physicians must
closely monitor renal function in patients on these drugs. Cox-2 inhibitors allegedly
produce less nephrotoxicity than do standard NSAID's. Limited experience exists with
the use of these agents to control chronic pain in people with sickle cell disease.
They are nonetheless worth serious consideration in these patients.
Opiod analgesics
Most patients who require large or frequent doses of opiods to control
pain are not seeking drugs for recreational purposes. Often, these patients
become tolerant to opiods. Consequently, the quantity of medication needed
to control severe pain exceeds that of an individual with a severe but
self-limited painful episode, such as torn knee ligaments. Most patients
report accurately the quantity of analgesics needed to control their pain
(Gil et al., 1994). In some instances, long-acting opiod analgesics can
blunt the severity of the pain, allowing patients to use less of the shorter
acting agents. Unfortunately, no objective measure of pain exists. Appropriate
treatment of pain irrespective of the cause requires an on-going dialogue
between the doctor and patient (Armstrong et al., 1992).
Whenever possible, patients should start taking their analgesics before
the pain becomes extremely severe. Maintaining pain at a tolerable level
is easier than reducing it from a high level of intensity. A typical episode
of severe sickle pain can require a patient to consume 4 to 8 mg of oral
hydromorphone every three hours to achieve relief. Many "severe" sickle
crises can be managed at home with analgesics, fluids, and rest. If the
pain progresses despite the use of reasonable quantities of medication,
the patient should seek emergency medical care.
Pain due to sickle cell disease is typically viewed as episodic
bouts secondary to occlusion of the microcirculation. Many health care
providers do not realize that severe chronic pain is also a consequence
of sickle cell disease. Chronic sickle cell pain occurs more commonly
in adults than in children. Permanent damage to the microcirculation secondary
to years of recurrent sickle injury likely is the basis of this syndrome.
Bony abnormalities on x-ray, such as vertebral body compression, attest
to the injury that occurs over the years. Other tissues, by inference,
suffer similar problems.
One of the obstacles to control of chronic pain is the short duration
of action common to many analgesics, such as hydromorphone and meperidine.
A number of longer acting formulations are available. One of the most effective
drugs for the control of chronic sickle cell pain is methadone. Although
best know for its use in narcotic detoxification programs, methadone is
a highly effective analgesic when given three times per day. Methadone
for control of narcotic addiction can be dispensed only at certified
detoxification
centers. Methadone for pain control can be given at other facilities in
accordance with the guidelines for use of any opiod.
Meperidine can present problems for pain control in patients with
sickle cell disease. The half-life of the drug in the circulation is
about 4 hours. The liver converts meperidine to normeperidine, a derivative
that has analgesic activity but which also is toxic. Grand mal seizure
is a particularly serious complication that occurs with the administration
of large amounts of meperidine. Normeperidine likely is the primary culprit
in this situation. Other opiod analgesics, therefore, are preferable to
meperidine. The American Pain Society recommends that meperidine no longer
be used for control of pain in people who require long-term analgesic treatment.
Drug-seeking behavior
Addiction is a concern for for medical providers and patients alike when chronic
pain require long-term use of opiods for control.
The magnitude of the problem is less than is often imagined, however. Those
who develop an addiction or drug-seeking behavior are seen frequently in
emergency rooms and as hospital inpatients. This overrepresentation of
a small number of patients in the health care system leads many providers
to conclude that drug-seeking behavior is a problem for most patients with
sickle cell disease. Opiods used for pain control, even when given in relatively
high doses, usually do not lead to addiction. As with any other group of
people, some patients with sickle cell disease have a propensity to develop
addictive disorders. The "easier access" that these patients have to narcotics
brings out a problem that might have developed in any event.
An important first step in managing this problem is to define drug-seeking
behavior. The use of large quantities of oral opiods or frequent visits to
the emergency room do not de facto signify drug-seeking behavior. Drug-seeking
behavior is the use of opiods in the absence of pain sufficiently severe
to justify these medications. Since no objective measure of pain exists,
reaching this conclusion is difficult. Drug-seeking behavior can be established
only by getting to know the patient and by observing the pattern of drug
usage. This means that over a period of months, frequent and heavy use
of opiods by the patient may need to be tolerated in order to establish
the pattern of drug consumption. Only then can the medical care provider
reasonably say on the basis of subtle signs such as facial expression,
vocal inflexion, pulse rate, etc., that drug-seeking behavior is likely.
At this point, the patient can be approached to discuss what appears to
be unwarranted use of drugs.
These patients can respond positively. Drug-seeking behavior can be
a very psychologically painful experience. Many patients are relieved when
they are confronted and given an option of help. Counseling or sessions
with a psychologist or social worker can be useful. In establishing drug-seeking
behavior, allowing a few patients to succeed in taking extra medication
for a while outweighs punishing patients who have a legitimate need by
placing arbitrary limits on everyone.
A few patients demonstrate incorrigible and sometimes frankly sociopathic
behavior. In these instances, the best approach is to limit opiod availability.
The patient should be informed that a limit is being imposed and the reason
for its implementation. Excuses for requesting additional medication fit
a pattern that can be a clue to drug-seeking behavior. Common pretexts
include, (a) forgetting to fill the prescription before expiration, (b)
losing the drug after the prescription is filled, (c) being robbed of the
prescription or the medication, (d) having a friend or relative abscond
with the medication after having the prescription filled. The limit should
be followed strictly. Tracking patterns of medication use is aided by keeping
a log of all opiod prescriptions issued. Photocopys of prescriptions provide
excellent documentation. If possible, a single provider at the institution
should write the prescriptions and maintain the record. Covering staff
and ER physicians should be alerted to the arrangement and should not supply
additional prescriptions. The arrangement is not punitive. Rather it allows
the staff to better assess and treat legitimate sickle cell pain. Health
providers should document their efforts to monitor and control excess use
of opiods by their patients. Scrupulous record-keeping also helps avoid
entanglements with medical practice oversight agencies.
Patients with extreme drug seeking behavior or sociopathic personality
disorders often acquire medication from other hospitals or medical facilities.
Sometimes this is done under the cover of an alias. Health-care providers
have limited ability to control such activity. The provider must be sure
that the pattern of drug use at his or her institution is reasonable, but
cannot police the entire region.
Support Groups/Psychiatric Support
The psychosocial dynamics of sickle cell disease are complex. As with any
other chronic, and often debilitating, illness, patients face a plethora
of social problems that greatly influence their clinical condition (Whitten
and Fischhoff, 1974). Loneliness, isolation, self-resentment, loss of
self-esteem,
and simple anger are common in patients with sickle cell disease. These
factors can profoundly influence the patient's ability to cope with pain.
Patient support groups and psychological counseling often are very useful.
The positive results in studies in which children were taught psychological
coping skills for pain, reinforce the importance of this component of patient
care (Gil et al., 1991).
Anemia
Vitamin Supplementation
Patients with sickle cell disease, like other people with hemolytic anemias,
require daily folic acid replacement. Folate is rapidly consumed by the
proliferating erythroid precursors. The normal daily intake of this vitamin
sometimes is insufficient to maintain a balance. One mg of supplemental
folate per day is more than enough to satisfy the needs of the erythron.
A patient with sickle cell disease whose hematocrit begins to fall unexpectedly
should be checked for folate deficiency as a part of the general work-up.
Sporadic Transfusion
Patients with sickle cell disease are anemic, by definition. The degree
of the anemia varies. The hematocrit frequently is in the mid- 20's. Some
patients have hematocrits in the low 30's while others have values in the
high teens. The baseline hematocrit remains relatively stable in a given
patient, however. Patients with Hemoglobin SC disease tend to run hematocrits
in the low to mid 30's. Most patients are conditioned to tolerate their
degree of anemia, and routine transfusion is not necessary. Raising the
hematocrit provides no clinical benefit, unless the baseline value has
fallen into the mid-teens, at which point oxygen carrying capacity can
be compromised. Hematocrits in such a low range leave little leeway for
further decline. On the other hand, transfusing patients with sickle cell
disease to hematocrits in the mid- to upper-30's can be dangerous, since
blood viscosity increases substantially at higher hematocrits (Kaul et
al., 1983). The increase in viscosity can worsen the sickling propensity
by increasing the time during which the cells remain in the low oxygen
tension regions of the circulation.
Chronic Transfusion Therapy
The best established use of chronic transfusion therapy is in patients
who have suffered strokes and have had initial exchange transfusions. Chronic
transfusion therapy is less well established for the treatment of other
complications of sickle cell disease. This modality has been advocated
as a means of treating recurrent severe episodes of sickle
pain, priapism, and as a prophylactic measure
inpregnant patients. Variable improvement in these
condition occurs. The utility of transfusion therapy is limited by
complications,
most notably alloimmunization and iron overload (Rosse et
al., 1990) (Wang et al., 1986) . Clinically
significant iron overload can occur after as few as 30 red cell transfusions.
The only treatment for transfusional iron overload
is chelation therapy with desferrioxamine
(Cohen and Schwartz, 1979). Marginal iron mobilization with this drug is
a frequent problem.
A major hurdle to the use of desferrioxamine is non-compliance. This
is a particular problem for young people, and occurs in other disorders
that require chelation therapy for transfusional iron overload, including
beta-thalassemia
and congenital sideroblastic anemia. Investigation of other chelators,
including some orally active drugs, is ongoing. Approval is imminent for
no agent, however.
Alloimmunization
Alloimmunization against minor red cell antigens is a major problem for
patients with sickle cell disease who receive frequent transfusions (Rosse
et al., 1990). The representation of minor antigens, such as Kell, Duffy,
and Kidd, differs between African-Americans and European- Americans (Issitt,
1994). For patients who receive only a few transfusions, the problem is
not serious. With repeated transfusion, however, antibodies develop against
these minor determinants complicating typing and jeopardizing further
transfusion.
Blacks are substantially underrepresented as blood donors, compounding
the problem of alloimmunization for patients with sickle cell disease.
In addition, patients with sickle cell disease appear to develop alloantibodies
more rapidly than other black patients who are transfused (Vichinsky et
al., 1990). Some institutions perform extended panel matching which includes
the most clinically significant minor antigens in an effort to delay the
development of alloantibodies. Some patients develop such severe problems
with alloantibodies that transfusion becomes nearly impossible. A number
of institutions have active programs to recruit blood donors from the black
community to lessen the impact of alloimmunization.
Routine use of blood from black donors for black patients with sickle
cell disease is not warranted. The likelihood of finding matched units
for patients with sickle cell disease is greater when black people are
in the donor pool. Matching is necessary nonetheless since antigen variation
among black people, like all other humans, is great. An expanded donor
pool substantially improves the chance of a match with antigen testing.
Age and severity of anemia
Sometimes, the severity of the anemia in patients with sickle cell disease
gradually increases as they age. The reason for this marrow "burn-out"
phenomenon is unknown. The clinical situation is complicated by the fact
that many of the patients have end-organ damage, such as a dilated
cardiomyopathy,
that may limit their ability to tolerate such severe anemia. Data from
the national cooperative study of sickle cell disease indicates that on
average patients with sickle cell disease survive until the mid 5th decade
of life. Bone marrow "burn-out" will be a greater issue as better general
medical care and new therapies prolong the lives of patients with sickle
cell disease.
Infection Prophylaxis
Infection is a leading cause of death in patients with sickle cell disease.
Hyposplenism, due to splenic autoinfarction, is a major contributor.
Hyposplenism
is not the sole cause of the defective host defense as evidenced by the
fact that overwhelming sepsis is the leading cause of death of children
under three years of age (Gill et al., 1995). Splenic autoinfarction is
less common in these very young children.
Antibiotics
A double-blind study of the use of penicillin prophylaxis for children
between the ages of six months and three years was terminated before the
expected time of completion (Gaston et al., 1986). The trend indicated
clearly that penicillin protected patients from infection or death due
to overwhelming infection by
Streptococcus pneumoniae. The recommendation
now is that all children be placed on prophylactic penicillin at a dose
of 250 mg twice a day. Patients with allergies to penicillin should be
treated with erythromycin. No recommended duration of treatment with
prophylactic
penicillin exists.
A second study looking at the role of prophylactic penicillin
in older children was recently completed. No difference in the incidence
of severe infection was found in this cohort of children between the ages
of 5 and 12 years (Falletta, et al., 1995). The implication is that penicillin
plays an important prophylactic role only in young children. One caveat
to the interpretation of this study is that the incidence of pneumoccocal
infection was strikingly low in both groups. This could have been a clinical
fluke. As such, a true difference in infection rate between the two groups
could possibly have been missed.
The role of prophylactic penicillin in adults with sickle cell
disease is unclear. Adults develop overwhelming sepsis, but at a much lower
frequency than do children. No controlled study to determine whether
prophylactic
antibiotics are useful in adults has been done. The recently completed
trial in older children suggest that prophylactic antibiotics may not benefit
adults. Nonetheless, many physicians still prescribe prophylactic antibiotics
for adults.
Immunization
Immunization with the pneumococcal vaccine is standard practice both in
adults and children with sickle cell disease. Several studies suggest that
immunization provides some protection, although incomplete, against pneumoccocal
infection (Ammann et al., 1977) (Ammann, 1982) (Schwartz, 1982). The vaccine
appears to be effective even in adults where splenic function has been
lost (Wong et al., 1992). The more recently available 23-valent vaccine
provides broader coverage than earlier versions. Although the duration
of protection is unknown, most specialists re-innoculate patients once
every 5 to 7 years. A noteworthy contrary voice comes from a broadbased
review of pneumococcal vaccine efficacy that cast doubt on the role of
the vaccine in patients with sickle cell disease (Butler, et al., 1993).
More recently, a vaccine against Hemophilus influenzae
has entered the clinical arena (Rubin et al., 1989). The efficacy of this
vaccine in sickle cell disease is unknown. Given the serious nature of
H.
influenzae infections in these patients, many specialists, particularly
pediatricians, now routinely immunize their patients against this organism.
Immunization against viral influenza is common practice. Viral
influenza per se is not a special threat for patients with sickle cell
disease. Since influenza is often complicated by bacterial infection and
other problems, prevention of the disease by immunization is a very practical
intervention.
Recently, an effective vaccine against hepatitis B was developed.
Since patients with sickle cell disease are likely to require one or more
transfusions in their lifetime, immunization against hepatitis B is a reasonable
precaution (Mok et al., 1989).
Avascular Necrosis of Bone
Avascular necrosis of bone is a common problem in patients with sickle
cell disease. This process is distinctly different from the acute bone
marrow necrosis discussed earlier. The areas most frequently affected are
cortical bone of the acetabulum, the head of the femur, and the head of
the humerus (Hernigou et al., 1993). The etiology of avascular necrosis
of bone is unknown. One hypothesis posits marrow hyperplasia in the femoral
head with tissue crowding and secondary reduced blood flow to the bone
as the inciting factor. Avascular necrosis also occurs in patients with
compound heterozygous conditions such as Hb SC disease, as well as in patients
with homozygous SS disease.
Patients usually report that the quality of the pain associated with
avascular bone necrosis differs substantially from their sickle cell pain.
The articular cartilage thins and often disappears as the process progresses.
The joints can deteriorate to a condition of bone-on-bone interface. Movement
of the joint becomes wrenchingly painful. Early on, non-steroidal
anti-inflammatory
agents can be useful. With more severe situations, particularly those that
involve the shoulder, injections of corticosteroids may help. Finally,
decompression of the tissue in the head of the humerus or the head of the
femur is used by some orthopedic surgeons with success. This invasive procedure
should be reserved for patients with more advanced cases of avascular necrosis.
The possible efficacy of femoral head core decompression is currently being
investigated in a multicenter study coordinated by Dr. Elliot Vichinsky
at the Children's Hospital of Oakland, California.
Even with these interventions, the process cannot be completely
halted, leading to joint replacement in some instances. Since most of the
patients are in their 20's or 30's when this becomes an issue, the decision
to proceed with joint replacement is difficult. Artificial joints are not
well-tolerated by some patients with sickle syndromes (Moran, 1995). As
many as one-third of patients require a second surgery within four years
of joint replacement. Also, these patients, for unclear reasons, are very
vulnerable to infections of their orthopedic hardware. The unfortunate
result sometimes is a destroyed articular interface and a flail joint which,
in the case of the femur, can leave the patient confined to a wheelchair.
More research is needed to identify patients at risk early in
the course of the degenerative process so that preventive measures can
be instituted. One promising addition to the diagnostic armamentarium is
MRI imaging. This technique can detect very early evidence of damage to
the bone, and holds the hope of improved management of this very debilitating
complication of sickle cell disease.
Osteomyelitis
Osteomyelitis often occurs at the site of necrotic segments of bone. Nearly
three-quarters of cases of osteomyelitis in patients with sickle cell disease
are due to Salmonella species (Anand and Glatt, 1994). Local pain and fever
are the most common indicators of chronic osteomyelitis (Epps Jr et al.,
1991). In the early stages of the disorder, bone roentenograms and even
bone scans frequently are unrevealing. Gallium scans can provide early
evidence of the disorder. Recently, MRI has been added to the diagnostic
arsenal, and appears to be a promising technique (Bonnerot et al., 1994).
Definitive diagnosis often requires bone biopsy. This procedure sometimes
is not an option, due to the location of the infection, however. Once the
diagnosis is made, four to six weeks of intravenous antibiotic therapy
are needed.
If a causative organism is not identified, broad- spectrum antibiotics
that have good tissue penetration should be used. Evidence of effectiveness,
in these instances, is an improvement in the pattern of fever and pain.
The advent of home infusion services obviates the need for prolonged
hospitalizations
in many cases.
Skin Ulcers
Skin ulcers are relatively infrequent in the United States in comparison
to reports from Jamaica. In that country, about 30% of patients with sickle
cell disease develop skin ulcers. This exceeds by far the incidence in
the US, which is closer to 1% (Koshy et al., 1989). Nonetheless, when skin
ulcers occur, the problems are very serious.
The most common site of skin ulcers is over the lateral malleoli. The
ulcerations often have no clear-cut antecedent trauma and progress over
a period of weeks to the point that the lesions extend into the dermis,
and often into the underlying subcutaneous tissue. With the breakdown in
the protection provided by the integument, patients are susceptible to
infections and other complications.
Treatment of ankle ulcers should be conservative. Rest, elevation, and
dry dressings with antimicrobial ointments are the best approaches to this
problem. Attempts at skin grafting are frequently thwarted by the poor
blood flow to the affected region. Healing usually takes weeks to months.
The area should be protected against trauma when the patient is up and
about (Wethers et al., 1994). Anecdotal reports exist of enhanced healing
of ulcers in patients placed on chronic transfusion therapy. In some instances,
patients were begun on chronic transfusion prior to skin grafting and maintained
with monthly transfusions for two or three months thereafter. Socks or
other clothing that cover the area should be avoided, to reduce friction
injury. A simple dry dressing provides additional protection. At one time,
a group of investigators advocated oral zinc supplementation as a means
of speeding the healing of ankle ulcers (Prasad et al., 1977). The rationale
was that patients with sickle cell disease are often deficient in zinc
(which is the second most common metal ion in red cells and is lost during
hemolysis). Zinc is important for wound healing. The evidence that zinc
supplementation aids the healing of ankle ulcers is controversial. However,
the benign nature of zinc supplementation makes it a reasonable option
in patients with this terribly debilitating and often recalcitrant condition.
Renal Dysfunction
The most common defect in patients with sickle cell disease is impaired
urine concentrating ability, or hyposthenuria (Kontessis, et al., 1992).
Hyposthenuria often occurs by the age of 3 or 4 years. The condition should
be considered in children with sickle cell disease who display bedwetting.
Hyposthenuria is seen in patients with homozygous sickle cell disease as
well as in compound heterozygotes (e.g., sickle-beta thalassemia). The
extremely high osmolality in the distal tubule produces some sickling even
of the cells in patients with sickle trait. As a consequence, hyposthenuria
is the most common abnormality seen that results from sickle cell trait (Gupta, et al., 1991).
The risk of renal dysfunction or failure is substantial in patients
with sickle cell disease (Flanagan et al., 1993). The high osmolality in
the renal medulla increases cell propensity to sickling. As a result, medullary
ischemia and papillary necrosis are common problems (Powars et al., 1991).
Sometimes, the necrotic papillae slough into the collecting system,
obstructing the outflow tract. No specific intervention has been devised
that is particularly effective in these patients. When the BUN and creatinine
begin to rise, limiting protein consumption is reasonable, as is recommended
for other patients with renal dysfunction. One report suggested that angiotensin
converting enzyme inhibitors may also retard the progression of nephropathy
in sickle cell disease (Falk et al., 1992). Further investigation of this
promising lead is needed.
Patients with sickle cell disease usually have low serum creatinine
and BUN levels. This is probably due to the high glomerular filtration
rate along with a high rate of creatinine secretion in the distal tubule.
BUN values of 7 and creatinine values of 0.5 are typical of those seen
in patients with sickle cell disease. A formal evaluation of glomerular
filtration rate should be considered in patients in whom the serum creatinine
rises above the level of about 1.0..
Potentially nephrotoxic drugs should be used with extreme caution
in patients with sickle cell disease. Antibiotics such as gentamicin should
be avoided when other agents are available that are less toxic to the kidney.
As noted previously, nonsteroidal anti-inflammatory drugs (NSAIDs) should
be used cautiously in patients with sickle cell disease. Heavy loads of
radiographic contrast agents formerly were a significant problem for patients
with sickle cell disease. The newer agents produce a much lower osmotic
load, with less dehydration of the kidneys, as a result.
Limited experience exists on the efficacy of dialysis in patients
with sickle cell disease (Falk et al., 1983). Reports that hemodialysis
is problematic in patients with sickle cell disease are anecdotal. Every
effort to prevent renal deterioration should be undertaken. Microscopic
hematuria is a common problem in patients with sickle cell disease (as
well as some patients with sickle cell trait). Hematuria per se requires
no intervention unless blood loss is massive. Some patients with sickle
cell disease and renal failure have received allografts (Gonzalez-Carrillo
et al., 1982).
People with sickle cell trait sometimes develop massive hematuria (Sears, 1978).
Interestingly,
the bleeding often comes from the left kidney. Hydration and alkalization
of the urine are commonly used interventions. Anecdotal reports of the
use of DDAVP in this situation are encouraging (Baldree, et al., 1990).
Episilon amino caproic acid (Amicar) has been used in some patients with
refractory bleeding from the kidney (Black, et al., 1976). Bleeding can
continue for weeks. Iron replacement may be necessary in some cases as
treatment interventions continue.
Retinopathy
Retinopathy is a significant problem
for 10% to 20% of patients with sickle cell disease (Moriarty et al., 1988).
The peak age of onset is in the 20's. For unknown reasons, the condition
develops more frequently in patients with hemoglobin SC disease than in
those with homozygous sickle cell disease (Clarkson, 1992). The retinopathy
resembles that seen in people with diabetes.
The condition is believed to result from ischemia to the retina.
The areas affected, at least initially, are in the periphery of the retina,
so that direct ophthalmoscopy is rarely revealing (Kimmel et al., 1986).
An ophthalmologist should evaluate the patient using pupillary dilation
and indirect ophthalmoscopy. Patients should be seen at least once a year,
and more frequently if abnormalities are noted. Ischemia can lead to retinal
thinning as well as neovascularization. The fragile vessels formed by
neovascularization
are subject to rupture, often leading to disastrous intraorbital hemorrhages.
This complication can produce sudden loss of vision (Pulido et al., 1988).
Laser photocoagulation has been used in an effort to prevent retinal hemorrhage
(Condon and Serjeant, 1980). Another common problem is retinal detachment,
particularly as a sequel to retinal hemorrhage.
Heart
Cardiomegaly is common in patients with sickle cell disease. Usually this
condition reflects sustained state of high cardiac output. While high output
failure occurs in some patients with sickle cell disease, the heart is
hyperdynamic in most (Gerry et al., 1978).
Pulmonary congestion due to fluid overload during hydration for painful
crisis is not a common problem in young patients. The picture changes as
patients age. A distinct minority of patients will develop problems with
fluid balance with the fluid challenge that occurs with hydration (Haynes,
Jr and Allison, 1986). For some patients, the problem is more one of left
atrial dysfunction than impaired ventricular activity. Nonetheless, patients
who develop a chest roentgenographic picture suggestive of "congestive
heart failure" during treatment for painful crisis must be examined carefully
to rule out other complications such as acute chest syndrome.
Pregnancy
Women with sickle cell disease can carry pregnancies to term, but the process
sometimes is complicated (Koshy et al., 1987) (Seoud et al., 1994). The
frequency of painful crises sometimes increases. Physical activity should
be limited, particularly after the mid-second trimester when the intravascular
volume increases substantially. Fetal development is usually normal if
the patient can be coaxed through the pregnancy (Powars et al., 1986).
Some specialists in sickle cell disease advocate simple blood transfusion
in these patients. Others have found no benefit to this intervention (Tuck
et al., 1987). The object is to maintain a hematocrit that allows normal
fetal development. Exchange transfusion has been used in some instances
in which patients had particularly difficult problems during the pregnancy.
These reports are anecdotal.
Women who have painful crises during pregnancy should be treated with
analgesics as necessary, including narcotics. The newborns who have been
exposed to opiods must be withdrawn by administering decreasing doses of
these drugs in the neonatal period. Warned of this issue, neonatologists
can easily manage the problem.
Newer Therapies
Therapies of Proven Benefit
Hydroxyurea
 Hydroxyureainhibits
ribonucleotide reductase, blocking DNA synthesis and cell division. The
drug also enhances fetal hemoglobin by developing erythroid cells (Platt
et al., 1984) (Stamatoyannopoulos and Nienhuis, 1992). Since fetal hemoglobin
blocks sickling, hydroxyurea has been administered to patients with sickle
cell disease in an effort to enhance fetal hemoglobin production (Charache,
1991) (Rodgers et al., 1990). Hydroxyurea induces fetal hemoglobin production,
increases the red cell mean corpuscular volume, and reduces the number
of dense cells and irreversibly sickled cells in the circulation (Goldberg
et al., 1992).
Hydroxyureainhibits
ribonucleotide reductase, blocking DNA synthesis and cell division. The
drug also enhances fetal hemoglobin by developing erythroid cells (Platt
et al., 1984) (Stamatoyannopoulos and Nienhuis, 1992). Since fetal hemoglobin
blocks sickling, hydroxyurea has been administered to patients with sickle
cell disease in an effort to enhance fetal hemoglobin production (Charache,
1991) (Rodgers et al., 1990). Hydroxyurea induces fetal hemoglobin production,
increases the red cell mean corpuscular volume, and reduces the number
of dense cells and irreversibly sickled cells in the circulation (Goldberg
et al., 1992).
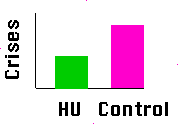 On
January 31, 1995, the Multicenter Study of Hydroxyurea in Sickle Cell Anemia
(MSH) was suspended by the NIH because patients on the hydroxyurea arm
of the study had significantly fewer painful crises than did the controls
(Charache et al., 1995). This made hydroxyurea the first (and only) drug
proven to prevent sickle cell crises. A second major observation was that
50% fewer episodes of acute chest syndrome occurred in the patients treated
with hydroxyurea. Hydroxyurea does not cure sickle cell disease, nor is
it effective in all patients. A detailed study showed that hydroxyurea
modifies the characteristics of red cells in patients with homozygous HbS
disease to resemble those of patients with HbSC disease (Bridges, et al,
1996). The heterogeneous response seen in the MSH study is consistent with
people with sickle cell disease being "converted" to a HbSC disease physiognomy.
Patients should be carefully screened and meet certain criteria:
On
January 31, 1995, the Multicenter Study of Hydroxyurea in Sickle Cell Anemia
(MSH) was suspended by the NIH because patients on the hydroxyurea arm
of the study had significantly fewer painful crises than did the controls
(Charache et al., 1995). This made hydroxyurea the first (and only) drug
proven to prevent sickle cell crises. A second major observation was that
50% fewer episodes of acute chest syndrome occurred in the patients treated
with hydroxyurea. Hydroxyurea does not cure sickle cell disease, nor is
it effective in all patients. A detailed study showed that hydroxyurea
modifies the characteristics of red cells in patients with homozygous HbS
disease to resemble those of patients with HbSC disease (Bridges, et al,
1996). The heterogeneous response seen in the MSH study is consistent with
people with sickle cell disease being "converted" to a HbSC disease physiognomy.
Patients should be carefully screened and meet certain criteria:
-
Age - 18 years or older.
-
Frequent painful vaso-occlusive crises. "Frequent" can reasonably be defined
as three or more crises per year that require hospitalization.
-
Use of accepted modes of contraception to prevent conception while on the
drug.
Hydroxyurea is not reasonable therapy for many patients with sickle cell
disease. Patients who have relatively few vaso-occlusive pain crises should
not receive hydroxyurea therapy. Other contraindications for hydroxyurea
include:
-
Pregnancy
-
Allergy to the drug
-
Thrombocytopenia or neutropenia
Although thrombocytopenia and/or neutropenia are relative contraindications,
some patients can tolerate the medication despite these pre-existing factors
with close monitoring. Bimonthly blood counts are required when patients
are started on hydroxyurea. In some patients on hydroxyurea, the hematocrit
rises to the high 30's or even low 40's. No evidence exists to support
hydroxyurea as prophylaxis against stroke, chronic leg ulcers, priapism,
or other complications of sickle cell disease.
The dose of hydroxyurea needed to prevent painful crises is unknown.
In the MSH study, patients received the maximum tolerated dose (MTD). The
dose administered was increased stepwise until signs of toxicity, such
as mild neutropenia, developed. The dose of hydroxyurea was then reduced
slightly. Whether such intense treatment is required is unknown.
Lower doses of hydroxyurea (e.g., 25mg/Kg/day) are used by some specialists.
Most patients treated with hydroxyurea develop macrocytosis (e.g., MCV=110).
Macrocytosis is not a good treatment gauge, however.
The data on hydroxyurea applies only to patients with homozygous sickle
cell disease (two hemoglobin S genes). Patients with compound heterozygous
conditions (e.g., sickle-beta thalassemia, Hb SC disease) were excluded
from the MSH study to eliminate if possible any response variability in
the data. Future trials may address these issues.
Hydroxyurea is not approved for use in children. The MSH study was restricted
to people 18 years of age or older. An NIH-sponsored trial of the drug
in children is on-going. A number of issues have been raised regarding
hydroxyurea in children, including neurocognitive development and bone
maturation. The pediatric hydroxyurea study will address some of these
issues.
Hydroxyurea is teratogenic in mice, but its toxicity to the human
fetus is unknown. The drug has not been associated with carcinogenesis.
The carcinogenic potential with very long-term use is unknown, however.
The NIH-sponsored Follow-up Study of Hydroxyurea in Sickle Cell Disease
is designed to monitor the 300 people in the original MSH study for long-term
side effects.
Bone Marrow Transplantation
Bone marrow
transplantation can
cure SCD. This intervention was first used in a patient with sickle cell
disease who also had relapsed acute lymphocytic leukemia. The transplant
was done to treat the leukemia, but cured the sickle cell disease as well.
The largest experience with transplantation for sickle cell disease comes
from Belgium and France, where about 80 patients have undergone bone marrow
transplantation (Apperley, 1993). The results have been quite promising
with cure of the sickle cell disease in every case in which engraftment
occurred.
Concerns about problems such as graft versus host disease and interstitial
pneumonia, two potentially fatal complications of bone marrow transplantation,
have limited the use of this modality in the United States (Kalinyak et
al., 1995) (Davies, 1993). A recently reported trial of bone marrow
transplantation
in children from centers in the US reaffirmed that the procedure can cure
sickle cell (Walters et al., 1996). Ten percent of the children died from
the procedure, and some suffered severe complications, such as graph rejection.
A later report by this group includes thirty-four children under the age
of 16 years who have received bone marrow transplants (Walters MC, et al.,
1997). The incidence of complications as lower in the children who underwent
transplant subsequent to the initial report.
The questions of when to perform a transplant and which patients
should receive the therapy are difficult. The optimal time for transplantation
is during childhood, since children fare better with transplantation than
do adults. The variable clinical manifestation of sickle cell disease makes
it impossible to predict in childhood which patients will have a more severe
course. This issue is particularly pertinent considering that transplantation
is best carried out prior to the development of end organ damage from the
sickle cell disease.
Analysis of data from the Study of the Natural History of Sickle Cell
Disease reported by Platt, et al, suggested that patients with fetal hemoglobin
levels of less than 8.6% tend to have more severe disease over the long
run (Platt et al., 1994). This would seem to provide a guide that could
be used in the decision of which patients to transplant. However, the data
are only statistical values. With rare exceptions, statistical data cannot
be applied to a particular patient to predict the clinical course.
An unresolved ethical question surrounds bone marrow transplantation
for children with sickle cell disease. Sickle cell disease is often a
debilitating
condition that makes life miserable for its victims. Although the Natural
History Study indicates that patients with sickle cell disease die earlier
than actuarial projections for other African-Americans, data collected
in the 1980's showed a life span that extends into the 40's. With the nearly
universal use of prophylactic penicillin in children to prevent overwhelming
pneumococcal infections along with other advances in supportive treatment,
this figure is likely to improve.
The question of who should decide to subject a child to this
potentially
fatal procedure likewise is complex. Should the decision be left to parents
and physicians? In a study at the University of Chicago, parents were presented
with hypothetical data on cure/mortality rates for their children with
sickle cell disease, and asked to indicate when the risks of the procedure
were acceptable relative to the gravity of the disease (Kodish et al.,
1991). About one-third of the parents indicated that a transplant mortality
rate of 15% along with a 15% incidence of graft-versus-host disease were
acceptable odds. However, young adults older than 18 years were not allowed
to participate in the decision process. Should the patients, the ones with
the most to gain and the most to lose, be excluded from the decision-making
loop? Should the courts appoint advocates for the children, to ensure that
the parents and physicians indeed are acting in the "best interest" of
the youngsters?
A program of bone marrow transplantation for beta-thalassemia major
has been successfully initiated in Italy (Lucarelli et al., 1993). Although
sickle cell disease and beta-thalassemia major are both hemoglobinopathies,
clear differences between the diseases exist. The most important is the
monotonous progression to disability and death that occurs with beta-thalassemia
major. Bone marrow transplantation for sickle cell disease offers promise.
The jury has not returned a final verdict, however.
Experimental Therapies
Erythropoietin
Erythropoietin is a hormone produced by the kidneys that stimulates red
cell production (Adamson and Eschbach, 1990). Usually, the hormone is made
in response to hypoxemia (Kario et al., 1992). Erythropoietin also increases
fetal hemoglobin levels in the red cells of many patients (Nagel et al.,
1993). A number of investigators have examined the extent to which
erythropoietin
will raise fetal hemoglobin levels in sickle cell disease. The consensus
is that the drug can significantly raise fetal hemoglobin levels, particularly
when given in high doses.
In one report, the drug was used in a dose of over 1,000 U/kg
three times per week (Rodgers et al., 1993). The treatment significantly
elevated fetal hemoglobin levels. The quantity of erythropoietin required
for this effect was enormous, particularly when compared to patients on
hemodialysis where the typical dose is now about 150 U/kg three times per
week. The cost of erythropoietin at the higher dose is prohibitive. Further,
while erythropoietin can increase the fetal hemoglobin content of red cells,
no controlled trial has shown that it alters the clinical course of sickle
cell disease.
Butyrate
Arginine butyrate and similar compounds have been tested in patients with
sickle cell disease (Perrine et al., 1989). The use of this agent stemmed
from the observation that babies born to diabetic mothers with poor glucose
control had sustained production of fetal hemoglobin after birth relative
to infants born to normal women. Butyrate produced as a byproduct of the
hyperglycemia produced the phenomenon. A group of investigators subsequently
examined the use of butyrate as a means of inducing fetal hemoglobin synthesis
in patients with sickle cells disease (Perrine et al., 1993).
Arginine butyrate can increase fetal hemoglobin levels, but the effect
is variable (Sher et al., 1995). Unfortunately, the drug must be given
intravenously and has a half-life of only about 5 minutes. Intermittent
rather than continuous infusion of arginine butyrate may induce fetal hemoglobin
synthesis more effectively. Side effects that have been seen in patients
who have received arginine butyrate include anorexia, nausea, vomiting,
and abnormal liver function tests (One patient had a seizure on the medication
after inadvertently receiving 4 times the recommended dose.) For most patients,
arginine butyrate is well-tolerated, however. The requirement for intravenous
administration limits the use of the agent. However, many drugs now are
administered intravenously to patients at home (e.g., desferrioxamine
for iron overload). Creative strategies are being explored to make arginine
butyrate a useful therapeutic option.
An effort is underway to identify orally active agents with longer
half-lives. Several compounds have been identified, and a couple have been
placed into early clinical trials. One of these is sodium phenylbutyrate,
a drug that has been used for patients with urea cycle disorders (Dover
et al., 1994). Unfortunately, patients tolerated the medication poorly,
in part due to the fact up to 40 tablets per day are needed to obtain acceptable
blood levels. The increase in Hb F levels produced by the oral agents studied
thus far have been significantly less than that seen with arginine butyrate
(Perrine et al., 1994). The butyrates have a number of hurtles to leap
before they are accepted for more general use, including a demonstration
that they consistently alter the red cell profile and, ultimately, improve
the clinical picture in sickle cell disease.
Clotrimazole
Red cell dehydration contributes
substantially to polymerization of sickle hemoglobin in patients with sickle
cell disease. The cell membrane is damaged in part through repeated physical
distortion by hemoglobin polymerization/depolymerization, and in part through
oxidant damage from reactive oxygen species generated by hemichromes and
other hemoglobin byproducts. K-CL co-transport increases, with K+
loss and associated water loss. Also, sickle cells accumulate Ca2+.
As a consequence, the Gardos channel (Ca2+-activated
K+ export) is activated, with further
dehydration.
Clotrimazole and other imidazole antimycotics specifically inhibit
the Ca2+-activated K+
channel pathway of normal and sickle erythrocytes. The original report
of Alvarez and colleagues described the inhibition of the normal human
red cell Gardos channel by clotrimazole (CLT) and other imidazole antimycotics.
Dr. Carlo Brugnara showed in sickle erythrocytes that CLT blocks K+
transport via the Gardos channel, prevents the change in membrane potential
observed when the Gardos channel is activated by internal Ca2+,
and inhibits dehydration induced by either the Ca2+
ionophore
A 23187 or cyclic oxygenation-deoxygenation.
Initial work by Dr. Brugnara and colleagues at Children's Hospital,
Boston showed that clotrimazole reduces the number of dense cells and the
number of irreversibly sickled cells in patients with sickle cell disease
(Brugnara, et al, 1996). A study of the agent at Children's Hospital
and Brigham and Women's Hospital evaluated the combined effects of clotrimazole
and hydroxyuea in patients with sickle cell disease. The hope was that the
agents, which have different mechanisms of action on sickle cells, would
work at least cooperatively, and perhaps synergistically, to reduce sickling. Clotrimazole
proved to have a number of side-effects that limited its success, including severe
dysuria in many men. A newer imidazole with fewer side-effects has recently come
available. Enrollment in the clotrimazole study has be suspended in anticipation
of this newer agent.
Nitric Oxide
Nitric oxide is one of the newest agents to enter testing for possible
treatment of patients with sickle cell disease. It is an inhaled gas that
has been used in a variety of investigational conditions, including neonatal
pulmonary hypertension and adult respiratory distress syndrome.
Nitric oxide is know primarily for its ability to relax smooth
muscle relaxation. However, the compound also forms a covalent link with
hemoglobin, particularly attaching as an S-nitroso group to the ß-93
cysteine (Gow and Stamler, 1998). This amino acid residue is near the "acceptor
pocket" on the ß-subunit of hemoglobin where the ß-6 valine
of Hb S forms a non-covalent interaction. The hydrophilic S-nitroso cysteine
at the ß-93 residue could destabilize the interaction between deoxy-Hb
S molecules in polymerized sickle hemoglobin.
Dr. C. Alvin Head, of Massachusetts General Hospital, and Dr.
Carlo Brugnara, of Children's Hospital, studied the interaction of nitric
oxide both in vitro and in vivo in normal volunteers and patients with
sickle cell disease (Head, et al., 1997). Their data suggested that nitric
oxide breathed at a concentration of 80 ppm reduces the polymerization
tendency of sickle hemoglobin. Reduced polymerization was inferred by a
fall in the P50 of sickle hemoglobin (no
effect occurred with Hb A). Nitric oxide for acute painful vaso-occlusive
crisis is being studied in an ongoing multicenter trial. Other investigators
were unable to reproduce the P50 effect
(Gladwin, et al., 1999). Further work is needed to determine whether nitric
oxide has a role in the treatment of patients with sickle cell disease.
FluocorTM
FluocorTM is a drug manufactured by the
CytRx Corporation of Norcross, Georgia. A phase III clinical trial of the
drug for patients with acute sickle cell pain crisis was recently completed.
FluocorTM is a more highly purified version
of the drug, RheothRxTM which went through
phase II clinical studies several years ago. Fifty patients were followed
in a placebo-control pilot study designed to evaluate safety and efficacy
of the compound (Adams-Graves, et al., 1997). The investigators infused
the drug continuously for 48 hours at the beginning of a sickle cell crisis.
The treated patients required less narcotic analgesic and showed a net
reduction in hospital length-of-stay relative to placebo control patients.
The multicenter study of FluocorTM
failed
to confirm these preliminary results. The future of the drug in the treatment
of sickle cell disease is unclear.
Gene Replacement Therapy
The beta-globin gene was
cloned a number of years ago, fueling interest in the possibility of gene
replacement therapy for sickle cell disease. While the idea of simply replacing
the defective gene with a normal one is appealing, a number of major
difficulties
must be surmounted. The first is to engineer a construct in which the
beta-globin
gene is expressed at high levels. Our understanding of the factors that
control globin gene expression has advanced significantly over the past
few years, but many of the nuances are yet to be worked out. A large segment
of DNA upstream of the beta-globin gene cluster, called the locus control
region, is necessary for efficient transcription of beta-globin mRNA. Any
attempt at gene therapy must include the large locus control region.
Another problem is that the inserted gene must be regulated in its expression
so that it produces beta-globin chains at a level roughly equal to the
production of endogenous alpha- globin chains. Failure to achieve such
a balance would produce a thalassemia. Further, the endogenous sickle
beta-globin
gene likely would have to be silenced, so that it does not continue to
produce sickle globin chains.
Finally, the cloned gene would have to be introduced into pluripotent
stem cells so that the patient would continue to make normal beta-globin
in perpetuity. The retroviral vectors that have been used to this point
infect dividing cells, while pluripotent stem cells divide very slowly.
To overcome this difficulty, a number of researchers have turned to
adeno-associated
viruses (AAV) as vehicles for gene therapy since these viruses can infect
resting cells. Here a new barrier, namely immune response to the viral
vector, has appeared. In any event, gene therapy for sickle cell disease,
the ultimate cure for the disorder, is not imminent.
Concluding Thoughts
Without major breakthroughs in gene therapy or bone marrow transplantation
that make these treatments applicable to a large number of patients, drug
intervention will remain the major therapeutic option for sickle cell disease.
The likelihood is low of finding a "magic bullet" medication that substantially
improves sickle cell disease for all or even most patients. Treatment likely
will involve the use of different agents alone or in combination to produce
optimal results. Most chronic illnesses, in fact, require combination drug
therapy. Hypertension, for instance, cannot be managed solely with diuretics.
Physicians test combinations of diuretics, beta blockers, calcium channel
blockers, and angiotensin converting enzyme (ACE) inhibitors to acheive
opitmal control of hypertension while producing the fewest side-effects.
Table 2. Future therapies for sickle cell disease
| Treatment Goal |
Cure |
Maintenance |
Acute Pain Management |
|
Bone Marrow Transplant |
hydroxyurea |
nitric oxide |
|
Gene Therapy |
clotrimazole |
FluocorTM |
|
|
magnesium pitolate |
Inibitiors of endothelial cell adhesion |
|
|
arginine butyrate |
Antiinflammatory agents |
Combination therapy currently is not an option for sickle cell disease,
since only hydroxyurea has been proven to alter the course of the condition.
Many, if not most of the agents currently under investigation likely will
fall short of investigators' hopes. If a few survive the rigors of testing
and joint the clinical armamentarium, however, we could mix and match drugs
for patients with sickle cell disease. Ideally, the treatment regimens
would include drugs with differing modes of action. Hydroxyurea, for instance,
combined with clotrimazole would team a drug that enhances fetal hemoglobin
production (hydroxyurea) with one that reduces erythrocyte dehydration
(clotrimazole). For a particular patient, sickle cell symptoms might be
improved substantially by neither drug alone. The combination, however,
might significantly ameliorate the condition.
Table 2 shows how therapies might be combined, using as examples some
of the agents currently under investigation. We cannot say whether the
therapeutic algorithm that eventually evolves will include these approaches
or modalities not yet conceived. The only statement that can be made with
confidence is that new vistas in the treatment of sickle cell disease will
usher in better and more fulfilling lives for these patients.
A major goal of investigation should be development of interventions
that can be used in very young patients. Many of the problems experienced
by adults and adolescents with sickle cell disease reflect incremental
organ damage by bouts of hypoxia. The affected areas may initially be
microscopic.
With time, these foci of injury colasce to form regions of macroscopic
injury, such as avascular necrosis of the femur. Prevention must be the
watchword as we seek to improve the management of patients with sickle
cell disease.
References
-
Adams, R., Mckie, V., Nichols, F., Carl, E., Zhang, D., Mckie, K., Figueroa,
R., Litaker, M., Thompson, W., and Hess, D. (1992). The use of transcranial
ultrasonography to predict stroke in sickle-cell disease. The New England
Journal of Medicine 326, 605-610.
-
Adams RJ, McKie VC, Brambilla D, Carl E, et. al. (1998). Stroke prevention
in sickle cell anemia. Control Clin Trials 19:110-129.
-
Adams-Graves P, Kedar A, Koshy M, Steinberg M, Veith R, et al. (1997) ReothRx
(poloxamer 188) injection for the acute painful episode of sickle cell
disease: a pilot study. Blood 90:2041-2046.
-
Adamson, J., and Eschbach, J. (1990). Treatment of anemia of chronic renal
failure with recombinant human erythropoietin. Annual Review of Medicine
41, 349-360.
-
Alvarez J , Montero M, Garcia-Sancho J: High affinity inhibition of
Ca2+-dependent
K+ channels by cytochrome P-450 inhibitors. J Biol Chem
1992,267:11789-11793.
-
Ammann, A. (1982). Current status of pneumococcal polysaccharide immunization
in patients with sickle cell disease or impaired splenic function. American
Journal of Pediatric Hematology Oncology 4, 301-6.
-
Ammann, A., Addiego, J., Wara, D., Lubin, B., Smith, W., and Mentzer, W.
(1977). Polyvalent pneumococcal-polysaccharide immunization of patients
with sickle-cell anemia and patients with splenectomy. New England Journal
of Medicine 297, 897-900.
-
Anand, A., and Glatt, A. (1994). Salmonella osteomyelitis and arthritis
in sickle cell disease. Semin Arthritis Rheum 24, 211-21.
-
Anastasi J. 1984. Hemoglobin S-mediated membrane oxidant injury: protection
from malaria and pathology in sickle cell disease. Med Hypotheses 14:
311-320.
-
Apperley, J. (1993). Bone marrow transplant for the haemoglobinopathies:
past, present and future. Baillieres Clin Haematol 6, 299-325.
Armstrong, F., Pegelow, C., Gonzalez, J., and Martinez, A. (1992).
Impact of children's sickle cell history on nurse and physician ratings
of pain and medication decisions. J Pediatr Psychol 17, 651-64.
-
Baldree LA, Ault BH, Chesney CM, Stapleton FB. (1990) Pediatr 86:238-243.
-
Balkaran, B., Char, G., Morris, J., Thomas, P., Serjeant, B., and Serjeant,
G. (1992). Stroke in a cohort of patients with homozygous sickle cell disease.
Journal of Pediatrics 120, 360-6.
-
Ballas, S. (1991). Sickle cell anemia with few painful crises is characterized
by decreased red cell deformability and increased number of dense cells.
Am J Hematol 36, 122-30.
-
Ballas, S., and Delengowski, A. (1993). Pain measurement in hospitalized
adults with sickle cell painful episodes. Ann Clin Lab Sci 23, 358-61.
-
Baruchel, S., Rees, J., Bernstein, M., and Goodyer, P. (1993). Relief of
sickle cell priapism by hydralazine. Report of a case. American Journal
of Pediatric Hematology-Oncology 15, 115-6.
-
Black WD, Hatch FE, Acchiardo S. Aminocaproic acid in prolonged hematuria
of patients with sicklemia. Arch Intern Med 1976; 136: 678-81.
-
Boletini, E., Svobodova, M., Divoky, V., Baysal, E., Curuk, M., Dimovski,
A., Liang, R., Adekile, A., and Huisman, T. (1994). Sickle cell anemia,
sickle cell beta-thalassemia, and thalassemia major in Albania: characterization
of mutations. Hum Genet 93, 182-7.
-
Bonnerot, V., Sebag, G., de Montalembert, M., Wioland, M., Glorion, C.,
Girot, R., and Lallemand, D. (1994). Gadolinium-DOTA enhanced MRI of painful
osseous crises in children with sickle cell anemia. Pediatr Radiol 24,
92-5.
-
Bouhassira, E., Lachman, H., Krishnamoorthy, R., Labie, D., and Nagel,
R. (1989). A gene conversion located 5' to the A gamma gene in linkage
disequilibrium with the Bantu haplotype in sickle cell anemia. Journal
of Clinical Investigation 83, 2070-2073.
-
Bridges KR, Garabino GD, Brugnara C, Cho MR, et al. (1996) A multiparameter
analysis of sickle erythrocytes in patients undergoing hydroxyurea therapy.
Blood 88:4701-4710.
-
Brugnara C, B Gee, C Armsby, S Kurth, M Sakamoto, N Rifai, SL Alper, O.
Platt: Therapy with oral clotrimazole induces inhibition of the Gardos
channel and reduction of erythrocyte dehydration in patients with sickle
cell disease. J. Clin. Invest. 1996; 97: 1227-1234.
-
Butler JC, Breiman RF, Campbell JF, Lipman HB, Broome CV, Facklam RR. (1993>
Pneumococcal polysaccharide vaccine efficacy. An evaluation of current
recommendations. JAMA 270 1826-1831.
-
Carlson, J., Nash, G., Gabutti, V., al-Yaman, F., and Wahlgren, M. (1994).
Natural protection against severe Plasmodium falciparum malaria due to
impaired rosette formation. Blood 84, 3909-3814. Charache, S. (1991).
Hydroxyurea
as treatment for sickle cell anemia. Hematol Oncol Clin North Am 5, 571-83.
-
Charache, S., Scott, J., and Charache, P. (1979). "Acute chest syndrome"
in adults with sickle cell anemia. Microbiology, treatment, and prevention.
Archives of Internal Medicine 139, 67-9.
-
Charache, S., Terrin, M., Moore, R., Dover, G., Barton, F., Eckert, S.,
McMahon, R., and Bonds, D. (1995). Effect of hydroxyurea on the frequency
of painful crises in sickle cell anemia. Investigators of the Multicenter
Study of Hydroxyurea in Sickle Cell Anemia. N Engl J Med 332, 1317-22.
-
Clark, M., Mohandas, N., and Shohet, S. (1980). Deformability of oxygenated
irreversibly sickled cells. J Clin Invest 65, 189-196.
-
Clarkson, J. (1992). The ocular manifestations of sickle-cell disease:
a prevalence and natural history study. Transactions of the American
Opthalmologic
Society 90, 481-504.
-
Cohen, A., Martin, M., Silber, J., Kim, H., Ohene-Frempong, K., and Schwartz,
E. (1992). A modified transfusion program for prevention of stroke in sickle
cell disease. Blood 79, 1657-61.
-
Cohen, A., and Schwartz, E. (1979). Iron chelation therapy in sickle cell
anemia. American Journal of Hematology 7, 69-76.
-
Condon, P., and Serjeant, G. (1980). Photocoagulation in proliferative
sickle retinopathy: results of a 5-year study. British Journal of Opthalmology
64, 832-40.
-
Davies, S. (1993). Bone marrow transplant for sickle cell disease--the
dilemma. Blood Reviews 7, 4-9.
-
Davies, S., and Brozovic, M. (1989). The presentation, management and
prophylaxis
of sickle cell disease. Blood Reviews 3, 29-44.
-
DeBaun, M., Glauser, T., Siegel, M., Borders, J., and Lee, B. (1995).
Noninvasive
central nervous system imaging in sickle cell anemia. A preliminary study
comparing transcranial Doppler with magnetic resonance angiography. J Pediatr
Hematol Oncol 17, 29-33.
-
Diamond, W., Brown Jr, F., Bitterman, P., Klein, H., Davey, R., and Winslow,
R. (1980). Delayed hemolytic transfusion reaction presenting as sickle-cell
crisis. Ann Intern Med 93, 231-4.
-
Dover, G., Brusilow, S., and Charache, S. (1994). Induction of fetal hemoglobin
production in subjects with sickle cell anemia by oral sodium phenylbutyrate.
Blood 84, 339-43.
-
Earley CJ, Kittner SJ, Feeser BR, Gardner J, Epstein A, Wozniak MA, Wityk
R, Stern BJ, Price TR, Macko RF, Johnson C, Sloan MA, Buchholz D. (1998)
Stroke in children and sickle-cell disease: Baltimore-Washington Cooperative
Young Stroke Study. Neurology 51:169-176.
-
Eaton, W., Hofrichter, J., and Ross, P. (1976). Delay Time of Gelation:
A Possible Determinant of Clinical Severity in Sickle Cell Disease. Blood
47, 621-627.
-
Eaton, W. A., and Hofrichter, J. (1990). Sickle cell hemoglobin polymerization.
[Review]. Advances in Protein Chemistry 40, 63-279.
-
Embury, S., Dozy, A., Miller, J., Davis, J. J., Kleman, K., Preisler, H.,
Vichinsky, E., Lande, W., Lubin, B., Kan, Y., and Mentzer, W. (1982). Concurrent
sickle-cell anemia and alpha-thalassemia: effect on severity of anemia.
N Engl J Med 306, 270-274.
-
Emond, A., Holman, R., Hayes, R., and Serjeant, G. (1980). Priapism and
impotence in homozygous sickle cell disease. Arch Intern Med 140, 1434-7.
-
Emre, U., Miller, S., Gutierez, M., Steiner, P., Rao, S., and Rao, M. (1995).
Effect of transfusion in acute chest syndrome of sickle cell disease. J
Pediatr 127, 901-4.
-
Epps Jr, C., Bryant 3d, D., Coles, M., and Castro, O. (1991). Osteomyelitis
in patients who have sickle-cell disease. Diagnosis and management. J Bone
Joint Surg 73, 1281-94.
-
Falk, R., Mattern, W., Lamanna, R., Gitelman, H., Parker, N., Cross, R.,
and Rastall, J. (1983). Iron removal during continuous ambulatory peritoneal
dialysis using deferoxamine. Kidney Int 24, 110-2.
-
Falk, R., Scheinman, J., Phillips, G., Orringer, E., Johnson, A., and Jennette,
J. (1992). Prevalence and pathologic features of sickle cell nephropathy
and response to inhibition of angiotensin-converting enzyme. The New England
Journal of Medicine 326, 910-5.
-
Fernbach, D., and Burdine Jr, J. (1970). Sepsis and functional asplenia.
New England Journal of Medicine 282, 691.
-
Flanagan, G., Packham, D., and Kincaid-Smith, P. (1993). Sickle cell disease
and the kidney. American Journal of Kidney Diseases 21, 325-7.
-
Fowler Jr, J., Koshy, M., Strub, M., and Chinn, S. (1991). Priapism associated
with the sickle cell hemoglobinopathies: prevalence, natural history and
sequelae. J Urol 145, 65-8.
-
Friedman, E., Webber, A., Osborn, H., and Schwartz, S. (1986). Oral analgesia
for treatment of painful crisis in sickle cell anemia. Ann Emerg Med 15,
787-91.
-
Gaston, M., Verter, J., Woods, G., Pegelow, C., Kelleher, J., Presbury,
G., Zarkowsky, H., Vinchinsky, E., Iyer, R., Lobel, J., Diamond, S., Holbrook,
C., Gill, F., Ritchey, K., Falletta, J., and Group, t. P. P. S. (1986).
Prophylaxis with oral penicillin in children with sickle cell anemia. A
randomized trial. New England Journal of Medicine 314, 1593-9.
-
Gelfand, M., Daya, S., Rucknagel, D., Kalinyak, K., and Paltiel, H. (1993).
Simultaneous occurrence of rib infarction and pulmonary infiltrates in
sickle cell disease patients with acute chest syndrome. Journal of Nuclear
Medicine 34, 614-8.
-
Gendrel, D., Kombila, M., Nardou, M., Gendrel, C., Djouba, F., and
Richard-Lenoble,
D. (1991). Protection against Plasmodium falciparum infection in children
with hemoglobin S. Pediatr Infect Dis J 10, 620-1.
-
Gerry, J., Bulkley, B., and Hutchins, G. (1978). Clinicopathologic analysis
of cardiac dysfunction in 52 patients with sickle cell anemia. Am J Cardiol
42, 211-6.
-
Gil, K., Phillips Jr, G., Edens, J., Martin, N., and Abrams, M. (1994).
Observation of pain behaviors during episodes of sickle cell disease pain.
Clin J Pain 10, 128-32.
-
Gil, K., Williams, D., Thompson, R., and Kinney, T. (1991). Sickle cell
disease in children and adolescents: the relation of child and parent pain
coping strategies to adjustment. J. Ped. Psych. 16, 643-663.
-
Gill, F., Sleeper, L., Weiner, S., Brown, A., Bellevue, R., Grover, R.,
Pegelow, C., and Vichinsky, E. (1995). Clinical events in the first decade
in a cohort of infants with sickle cell disease. Cooperative Study of Sickle
Cell Disease. Blood 86, 776-83.
-
Gillams AR, McMahon L, Weinberg G, Carter AP (1998) MRA of the intracranial
circulation in asymptomatic patients with sickle cell disease. Pediatr
Radiol 28:283-287.
-
Gladwin MT, Schechter AN, Shelhamer JH, Pannell LK, Conway DA, Hrinczenko
BW, Nichols JS, Pease-Fye ME, Noguchi CT, Rodgers GP, Ognibene FP. (1999)
Inhaled nitric oxide augments nitric oxide transport on sickle cell hemoglobin
without affecting oxygen affinity. J Clin Invest 104:937-945
-
Goldberg, M., Brugnara, C., Dover, G., Schapira, L., Charache ,S., and
Bunn, H. (1990. Treatment of sickle cell anemia with hydroxyurea and
erythropoietin.
N Eng J Med 323, 366-372.
-
Goldberg, M., Brugnara, C., Dover, G., Schapira, L., Lacroix, L., and Bunn,
H. (1992). Hydroxyurea and erythropoietin therapy in sickle cell anemia.
Seminarys in Oncology 19, 74-81.
-
Gonzalez-Carrillo, M., Rudge, C., Parsons, V., Bewick, M., and White, J.
(1982). Renal transplantation in sickle cell disease. Clin Nephrol 18,
209-10.
-
Gow AJ, Stamler JS. (1998) Reactions between nitric oxide and haemoglobin
under physiological conditions. Nature 391:169-73.
-
Griffin, T., McIntire, D., and Buchanan, G. (1994). High-dose intravenous
methylprednisolone therapy for pain in children and adolescents with sickle
cell disease. N Engl J Med 330, 733-737.
-
Gupta AK, Kirchner KA, Nicholson R, Adams JG 3d, Schechter AN, Noguchi
CT, Steinberg MH. (1991) Effects of alpha-thalassemia and sickle polymerization
tendency on the urine-concentrating defect of individuals with sickle cell
trait J Clin Invest 88: 1963-1968.
-
Haynes Jr, J., and Allison, R. (1986). Pulmonary edema. Complication in
the management of sickle cell pain crisis. Am J Med 80, 833-40.
-
Haynes Jr, J., and Kirkpatrick, M. (1993). The acute chest syndrome of
sickle cell disease. American Journal of Medical Science 305, 326-30.
-
Head CA, Brugnara C, Martinez-Ruiz R, Kacmarek, et al. (1997) Low concentrations
of nitric oxide increase oxygen affinity of sickle erythrocytes in vitro
and in vivo. J Clin Invest 100:1193-1198.
-
Hernigou, P., Bachir, D., and Galacteros, F. (1993). Avascular necrosis
of the femoral head in sickle-cell disease. Treatment of collapse by the
injection of acrylic cement. J Bone Joint Surg Br 75, 875-80.
-
Holbrook, C. (1990). Patient-controlled analgesia pain management for children
with sickle cell disease. J Assoc Acad Minor Phys 1, 93-6.
-
Humbert, J., Winsor, E., Githens, J., and Schmitz, J. (1990). Neutrophil
dysfunctions in sickle cell disease. Biomed Pharmacother 44, 153-8.
-
Ingram, V.M. (1956) A specific chemical difference between globins of normal
and sickle-cell anúmia húmoglobins. Nature 178, 792-794.
-
Issitt, P. (1994). Race-related red cell alloantibody problems. Br J Biomed
Sci 51, 158-67.
-
Johnson, K., Stastny, J., and Rucknagel, D. (1994). Fat embolism syndrome
associated with asthma and sickle cell-beta(+)-thalassemia. Am J Hematol
46, 354-7.
-
Kalinyak, K., Morris, C., Ball, W., Ris, M., Harris, R., and Rucknagel,
D. (1995). Bone marrow transplantation in a young child with sickle cell
anemia. Am J Hematol 48, 256-61.
-
Kar, B., Satapathy, R., Kulozik, A., Kulozik, M., Sirr, S., Serjeant, B.,
and Serjeant, G. (1986). Sickle cell disease in Orissa State, India. Lancet
2, 1198-201.
-
Kario, K., Matsuo, T., Kodama, K., Nakao, K., and Asada, R. (1992). Reduced
erythropoietin secretion in senile anemia. Am J Hematol 41, 252-7.
-
Kaul, D., Fabry, M., and Nagel, R. (1989). Microvascular sites and
characteristics
of sickle cell adhesion to vascular endothelium in shear flow conditions:
pathophysiological implications. Proc Natl Acad Sci U S A 86, 3356-60.
-
Kaul, D. K., Fabry, M. E., Windisch, P., Baez, S., and Nagel, R. L. (1983).
Erythrocytes in sickle cell anemia are heterogeneous in their rheological
and hemodynamic characteristics. Journal of Clinical Investigation 72,
22-31.
-
Keidan, A., Marwah, S., Vaughan, G., Franklin, I., and Stuart, J. (1987).
Painful sickle cell crises precipitated by stopping prophylactic exchange
transfusions. J Clin Pathol 40, 505-7.
-
Kimmel, A., Magargal, L., and Tasman, W. (1986). Proliferative sickle
retinopathy
and neovascularization of the disc: regression following treatment with
peripheral retinal scatter laser photocoagulation. Ophthalmic Surgery 17,
20-2.
-
Kinney, T., Ware, R., Schultz, W., and Filston, H. (1990). Long-term
management
-
Kinney TR, Sleeper LA, Wang WC, Zimmerman RA, Pegelow CH, Ohene-Frempong
K, Wethers DL, Bello JA, Vichinsky EP, Moser FG, Gallagher DM, DeBaun MR,
Platt OS, Miller ST. (1999) Silent cerebral infarcts in sickle cell anemia:
a risk factor analysis. The Cooperative Study of Sickle Cell Disease. Pediatrics
103:640-645.
of splenic sequestration in children with sickle cell disease. J Pediatr
117, 194-9.
-
Kirkpatrick, M., Haynes Jr, J., and Bass Jr, J. (1991). Results of
bronchoscopically
obtained lower airway cultures from adult sickle cell disease patients
with the acute chest syndrome. American Journal of Medicine 90, 206-10.
-
Kodish, E., Lantos, J., Stocking, C., Singer, P., Siegler, M., and Johnson,
F. (1991). Bone marrow transplantation for sickle cell disease. A study
of parent's decisions. N Engl J Med 325, 1349-1353.
-
Kontessis P, Mayopoulou-Symvoulidis D, Symvoulidis A, Kontopoulou-Griva
I. (1992) Renal involvement in sickle cell-beta thalassemia. Nephron 61:
10-15.
-
Koren, A., Wald, I., Halevi, R., and Ben Ami, M. (1990). Acute chest syndrome
in children with sickle cell anemia. Pediatric Hematology Oncology 7,
99-107.
-
Koshy, M., Burd, L., Dorn, L., and Huff, G. (1987). Frequency of pain crisis
during pregnancy. Prog Clin Biol Res 240, 305-11.
-
Koshy, M., Entsuah, R., A, K., Kraus, A., Johnson, R., Bellvue, R.,
Flournoy-Gill,
Z., and Levy, P. (1989). Leg ulcers in patients with sickle cell disease.
Blood 74, 1403-8.
-
Landesman, S., Rao, S., and Ahonkhai, V. (1982). Infections in children
with sickle cell anemia. Special reference to pneumococcal and salmonella
infections. American Journal of Pediatric Hematology and Oncology 4,
407-18.
-
Lanzkowsky, P., Shende, A., Karayalcin, G., Kim, Y., and Aballi, A. (1978).
Partial exchange transfusion in sickle cell anemia. Use in children with
serious complications. Am J Dis Child 132, 1206-8.
-
Lucarelli, G., Galimberti, M., Polchi, P., Angelucci, E., Baronciani, D.,
Giardini, C., Andreani, M., Agostinelli, F., Albertini, F., and Clift,
R. (1993). Marrow transplantation in patients with thalassemia responsive
to iron chelation therapy. N Engl J Med 329, 840-844.
-
Mallouh, A., and Qudah, A. (1993). Acute splenic sequestration together
with aplastic crisis caused by human parvovirus B19 in patients with sickle
cell disease. Journal of Pediatrics 122, 593-5.
-
Mankad, V., Williams, J., Harpen, M., Manci, E., Longenecker, G., RB, M.,
Shah, A., Yang, Y., and Brogdon, B. (1990). Magnetic resonance imaging
of bone marrow in sickle cell disease: clinical, hematologic, and pathologic
correlations. Blood 75, 274-83.
- Mantadakis E, Cavender JD, Rogers ZR, Ewalt DH, Buchanan GR. 1999.
Prevalence of priapism in children and adolescents with sickle cell anemia. J
Pediatr Hematol Oncol 21: 518-522.
-
McPherson, E., Perlin, E., Finke, H., Castro, O., and Pittman, J. (1990).
Patient-controlled analgesia in patients with sickle cell vaso-occlusive
crisis. Am J Med Sci 299, 10-2.
-
Miller, B., Olivieri, N., Salameh, M., Ahmed, M., Antognetti, G., Huisman,
T., Nathan, D., and Orkin, S. (1987). Molecular analysis of the
high-hemoglobin-F
phenotype in Saudi Arabian sickle cell anemia. N Engl J Med 316, 244-50.
-
Miller, S., Hammerschlag, M., Chirgwin, K., Rao, S., Roblin, P., Gelling,
M., Stilerman, T., Schachter, J., and Cassell, G. (1991). Role of Chlamydia
pneumoniae in acute chest syndrome of sickle cell disease. Journal of Pediatrics
118, 30-3.
- Miller ST, Sleeper, LA, Pegelow CH, Enos LE, Wang WC, Weiner SJ, Wethers DL,
Smith J, Kinney TR. (2000) Prediction of adverse outcomes in children with
sickle cell disease.
N Engl J Med 342: 83-99.
-
Mok, Q., Underhill, G., Wonke, B., Aldouri, M., Kelsey, M., and Jefferies,
D. (1989). Intradermal hepatitis B vaccine in thalassaemia and sickle cell
disease. Arch Dis Child 64, 535-40.
-
Moran, M. (1995). Osteonecrosis of the hip in sickle cell hemoglobinopathy.
Am J Orthop 24, 18-24.
-
Moriarty, B., Acheson, R., Condon, P., and Serjeant, G. (1988). Patterns
of visual loss in untreated sickle cell retinopathy. Eye 2, 330-5.
-
Mykulak, D., and Glassberg, K. (1990). Impotence following childhood priapism.
J Urol 144, 134-5.
-
Nagel, R., and Fleming, A. (1992). Genetic epidemiology of the ßS
gene. Baillieres Clin Haematol 5, 331-365.
-
Nagel, R., Vichinsky, E., Shah, M., Johnson, R., Spadacino, E., Fabry,
M., Mangahas, L., Abel, R., and Stamatoyannopoulos, G. (1993). F reticulocyte
response in sickle cell anemia treated with recombinant human erythropoietin:
a double-blind study. Blood 81, 9-14.
-
Ohene-Frempong, K. (1991). Stroke in sickle cell disease: demographic,
clinical and therapeutic considerations. Seminars in Hematology 28,
213-219.
- Olatunji PO, Davies SC. (2000) The predictive value of white cell count in
assessing clinical severity of sickle cell anaemia in Afro-Caribbeans patients.
Afr J Med Med Sci 29: 27-30.
-
Olopoenia, L., Frederick, W., Greaves, W., Adams, R., Addo, F., and Castro,
O. (1990). Pneumococcal sepsis and meningitis in adults with sickle cell
disease. Southern Medical Journal 83, 1002-4.
-
Orjih, A., Chevli, R., and Fitch, C. (1985). Toxic heme in sickle cells:
an explanation for death of malaria parasites. Am J Trop Med Hyg 34, 223-7.
-
Overturf, G., Powars, D., and Baraff, L. (1977). Bacterial meningitis and
septicemia in sickle cell disease. American Journal of Diseases of Childhood
131, 784-7.
-
Pegelow, C. (1992). Survey of pain management therapy provided for children
with sickle cell disease. Clin Pediatr (Phila) 31, 211-4.
-
Pegelow, C., Adams, R., McKie, V., Abboud, M., Berman, B., Miller, S.,
Olivieri, N., Vichinsky, E., Wang, W., and Brambilla, D. (1995). Risk of
recurrent stroke in patients with sickle cell disease treated with erythrocyte
transfusions. J Pediatr 126, 896-9.
-
Perlin, E., Finke, H., Castro, O., Rana, S., Pittman, J., Burt, R., Ruff,
C., and McHugh, D. (1994). Enhancement of pain control with ketorolac
tromethamine
in patients with sickle cell vaso-occlusive crisis. Am J Hematol 46, 43-7.
-
Perrine, R., Pembrey, M., John, P., Perrine, S., and Shoup, F. (1978).
Natural history of sickle cell anemia in Saudi Arabs. A study of 270 subjects.
Ann Intern Med 88, 1-6.
-
Perrine, S., Dover, G., Daftari, P., Walsh, C., Jin, Y., Mays, A., and
Faller, D. (1994). Isobutyramide, an orally bioavailable butyrate analogue,
stimulates fetal globin gene expression in vitro and in vivo. Br J Haematol
88, 555-61.
-
Perrine, S., Ginder, G., Faller, D., Dover, G., Ikuta, T., Witkowska, E.,
Cai, S.-P., Vichinsky, E., and Olivieri, N. (1993). A short-term trial
of butyrate to stimulate fetal-globin-gene expression in the beta-globin
disorders. New England Journal of Medicine 328, 81-86.
-
Perrine, S., Miller, B., Faller, D., Cohen, R., Vichinsky, E., Hurst, D.,
Lubin, B., and Papayannopoulou, T. (1989). Sodium butyrate enhances fetal
globin gene expression in erythroid progenitors of patients with Hb SS
and beta thalassemia. Blood 74, 454-9.
-
Platt, O., Brambilla, D., Rosse, W., Milner, P., Castro, O., Steinberg,
M., and Klug, P. (1994). Mortality in sickle cell disease. Life expectancy
and risk factors for early death. N Engl J Med 330, 1639-44.
-
Platt, O., Orkin, S., Dover, G., Beardsley, G., Miller, B., and Nathan,
D. (1984). Hydroxyurea enhances fetal hemoglobin production in sickle cell
anemia. J Clin Invest 74, 652-656.
-
Platt, O., Thorington, B., Brambilla, D., Milner, P., Rosse, W., Vichinsky,
E., and Kinney, T. (1991). Pain in sickle cell disease. Rates and risk
factors. N Engl J Med 325, 11-16.
-
Poncz, M., Kane, E., and Gill, F. (1985). Acute chest syndrome in sickle
cell disease: etiology and clinical correlates. Journal of Pediatrics 107,
861-6.
-
Powars, D., Elliott-Mills, D., Chan, L., Niland, J., Hiti, A., Opas, L.,
and Johnson, C. (1991). Chronic renal failure in sickle cell disease: risk
factors, clinical course, and mortality. Ann Intern Med 115, 614-20.
-
Powars, D., and Hiti, A. (1993). Sickle cell anemia. Beta s gene cluster
haplotypes as genetic markers for severe disease expression. Am J Dis Child
147, 1197-1202.
-
Powars, D., Sandhu, M., Niland-Weiss, J., Johnson, C., Bruce, S., and Manning,
P. (1986). Pregnancy in sickle cell disease. Obstet Gynecol 67, 217-28.
-
Powars DR, Conti PS, Wong WY, Groncy P, Hyman C, Smith E, Ewing N, Keenan
RN, Zee CS, Harold Y, Hiti AL, Teng EL, Chan LS. (1999) Cerebral vasculopathy
in sickle cell anemia: diagnostic contribution of positron emission tomography.
Blood 93:71-79.
-
Prasad, A., Abbasi, A., and Ortega, J. (1977). Zinc deficiency in man:
studies in sickle cell disease. Prog Clin Biol Res 14, 211-239.
-
Pulido, J., Flynn Jr, H., Clarkson, J., and Blankenship, G. (1988). Pars
plana vitrectomy in the management of complications of proliferative sickle
retinopathy. Arch Ophthalmol 106, 1553-7.
-
Ramasamy, S., Balakrishnan, K., and Pitchappan, R. (1994). Prevalence of
sickle cells in Irula, Kurumba, Paniya & Mullukurumba tribes of Nilgiris
(Tamil Nadu, India). Indian J Med Res 100, 242-5.
-
Rao, V., Mitchell, D., Rifkin, M., Steiner, R., Burk Jr, D., Levy, D.,
and Ballas, S. (1989). Marrow infarction in sickle cell anemia: correlation
with marrow type and distribution by MRI. Magnetic Resonance Imaging 7,
39-44. <
-
Robieux, I., Kellner, J., Coppes, M., Shaw, D., Brown, E., Good, C.,
O'Brodovich,
H., Manson, D., Olivieri, N., Zipursky, A., and al, e. (1992). Analgesia
in children with sickle cell crisis: comparison of intermittent opioids
vs. continuous intravenous infusion of morphine and placebo-controlled
study of oxygen inhalation. Pediatric Hematology Oncology 9, 317-26.
-
Rodgers, G., Dover, D., Uyesaka, N., Noguchi, C., Schechter, A., and Neinhuis,
A. (1993). Augmentation by erythropoietin of the fetal-hemoglobin response
to hydroxyurea in sickle cell disease. New England Journal of Medicine
328, 73-80.
-
Rodgers, G., Dover, G., Noguchi, C., Schechter, A., and Nienhuis, A. (1990).
Hematologic responses of patients with sickle cell disease to reatment
with hydroxyurea. N Engl J Med 322, 1037-45.
-
Roshkow, J., and Sanders, L. (1990). Acute splenic sequestration crisis
in two adults with sickle cell disease: US, CT, and MR imaging findings.
Radiology 177, 723-5.
-
Rosse, W., Gallagher, D., Kinney, T., Castro, O., Dosik, H., Moohr, J.,
Wang, W., and PS, L. (1990). Transfusion and alloimmunization in sickle
cell disease. The Cooperative Study of Sickle Cell Disease. Blood 76,
1431-7.
- Roth Jr, EF, Friedman M, Ueda Y, Tellez I, Trager W Nagel RL. 1978. Sickling
rates of human AS red cells infected in vitro with Plasmodium falciparum
malaria. Science 202: 650-652.
-
Rubin, L., Voulalas, D., and Carmody, L. (1989). Immunization of children
with sickle cell disease with Haemophilus influenzae type b polysaccharide
vaccine. Pediatrics 84, 509-13.
-
Saarinen, U., Chorba, T., Tattersall, P., Young, N., Anderson, L., Palmer,
E., and Coccia, P. (1986). Human parvovirus B19-induced epidemic acute
red cell aplasia in patients with hereditary hemolytic anemia. Blood 67,
1411-7.
-
Sanders, D., Severance, H., and Pollack, C. J. (1992). Sickle cell
vaso-occlusive
pain crisis in adults: alternative strategies for management in the emergency
department. Southern Medical Journal 85, 808-11.
-
Schiliro, G., Spena, M., Giambelluca, E., and Maggio, A. (1990). Sickle
hemoglobinopathies in Sicily. American Journal of Hematology 33, 81-5.
-
Schwartz, J. (1982). Pneumococcal vaccine: clinical efficacy and effectiveness.
Annals of Internal Medicine 96, 208-20.
-
Sears D. (1978) The morbidity of sickle cell trait: a review of the literature.
Am J Med 64:1021-1036.
-
Sears, D., and Udden, M. (1985). Splenic infarction, splenic sequestration,
and functional hyposplenism in hemoglobin S-C disease. American Journal
of Hematology 18, 261-8.
-
Seeler, R. (1973). Intensive transfusion therapy for priapism in boys with
sickle cell anemia. J Urol 110, 360-3.
-
Seoud, M., Cantwell, C., Nobles, G., and Levy, D. (1994). Outcome of pregnancies
complicated by sickle cell and sickle-C hemoglobinopathies. Am J Perinatol
11, 187-91.
-
Serjeant, G., de Ceulaer, K., and Maude, G. (1985). Stilboestrol and stuttering
priapism in homozygous sickle-cell disease. Lancet 2, 1274-6.
-
Shapiro, M., and Hayes, J. (1984). Fat embolism in sickle cell disease.
Report of a case with brief review of the literature. Archives of Internal
Medicine 144, 181-2.
-
Sher, G., Ginder, G., Little, J., Yang, S., Dover, G., and Olivieri, N.
(1995). Extended therapy with intravenous arginine butyrate in patients
with beta-hemoglobinopathies. N Engl J Med 332, 1606-10.
-
Solanki, D., Kletter, G., and Castro, O. (1986). Acute splenic sequestration
crises in adults with sickle cell disease. American Journal of Medicine
80, 985-90.
-
Sprinkle, R., Cole, T., Smith, S., and Buchanan, G. (1986). Acute chest
syndrome in children with sickle cell disease. A retrospective analysis
of 100 hospitalized cases. American Journal of Pediatric Hematology Oncology
8, 105-10.
-
Stamatoyannopoulos, G., Wood, W., Papayannopoulou, T., and Nute, P. (1975).
A new form of hereditary persistence of fetal hemoglobin in blacks and
its association with sickle cell trait. Blood 46, 683-92.
-
Stamatoyannopoulos, J., and Nienhuis, A. (1992). Therapeutic approaches
to hemoglobin switching in treatment of hemoglobinopathies. Annu Rev Med
43, 497-521.
-
Steinberg, M., and Embury, S. (1986). Alpha-thalassemia in blacks: genetic
and clinical aspects and interactions with the sickle hemoglobin gene.
Blood 68, 985-90.
-
Szwed, J., Yum, M., and Hogan, R. (1980). A beneficial effect of splenectomy
in sickle cell anemia and chronic renal failure. Am J Med Sci 279, 169-72.
-
Tobias, J. (1993). Indications and application of epidural anesthesia in
a pediatric population outside the perioperative period. Clin Pediatr (Phila)
32, 81-5.
-
Tuck, S., James, C., Brewster, E., Pearson, T., and Studd, J. (1987).
Prophylactic
blood transfusion in maternal sickle cell syndromes. Br J Obstet Gynaecol
94, 121-5.
-
Van Hoff, J., Ritchey, A., and Shaywitz, B. (1985). Intracranial hemorrhage
in children with sickle cell disease. Am J Dis Child 139, 1120-3.
-
Vichinsky, E., Earles, A., Johnson, R., Hoag, M., Williams, A., and Lubin,
B. (1990). Alloimmunization in sickle cell anemia and transfusion of racially
unmatched blood. N Engl J Med 322, 1617-21.
-
Vichinsky, E., and Lubin, B. (1980). Sickle cell anemia and related
hemoglobinopathies.
Pediatr Clin North Am 27, 429-447.
-
Vichinsky, E., Williams, R., Das, M., Earles, A., Lewis, N., Adler, A.,
and McQuitty, J. (1994). Pulmonary fat embolism: a distinct cause of severe
acute chest syndrome in sickle cell anemia. Blood.
-
Walters, M., Patience, M., Leisenring, W., Eckman, J., Scott, J., Mentzer,
W., Davies, S., Ohene-Frempong, K., Bernaudin, F., Matthews, D., Storb,
R., and Sullivan, K. (1996). Bone marrow transplantation for sickle cell
disease. N Engl J Med 335, 369-376.
-
Walters MC, Patience M, Leisering W, Rogers ZR, et. al. (1997) Collaborative
multicenter investigation of marrow transplantation for sickle cell disease:
current results and future directions. Biol Blood Marrow Transplant
3:310-315.
-
Wang, W., Ahmed, N., and Hanna, M. (1986). Non-transferrin-bound iron in
long-term transfusion in children with congenital anemias. J Pediatr 108,
552-557.
-
Wang, W., Kovnar, E., Tonkin, I., Mulhern, R., Langston, J., Day, S., Schell,
M., and Wilimas, J. (1991). High risk of recurrent stroke after discontinuance
of five to twelve years of transfusion therapy in patients with sickle
cell disease. J Pediatr 118, 377-82.
-
Wang, Z., Bogdan, A., Zimmerman, R., Gusnard, D., Leigh, J., and Ohene-Frempong,
K. (1992). Investigation of stroke in sickle cell disease by 1H nuclear
magnetic resonance spectroscopy. Neuroradiology 35, 57-65.
Wethers, D. (1982). Problems and complications in the adolescent with
sickle cell disease. Am J Pediatr Hematol Oncol 4, 47-53.
-
Wethers, D., Ramirez, G., Koshy, M., Steinberg, M., Phillips Jr., G., Siegel,
R., Eckman, J., and Prchal, J. (1994). Accelerated healing of chronic
sickle-cell
leg ulcers treated with RGD peptide matrix. RGD Study Group. Blood 84,
1775-9.
-
White DA, DeBaun M. (1998) Cognitive and behavioral function in children
with sickle cell disease: a review and discussion of methodological issues.
J Pediatr Hematol Oncol 20:458-462.
- Willcox M, Bjorkman A, Brohult J, Pehrson PO, Rombo L, Bengtsson E. 1983. A
case-control study in northern Liberia of Plasmodium
falciparum malaria in haemoglobin S and beta-thalassaemia traits. Ann
Trop Med Parasitol 77:239-246.
-
Wong, W., Powars, D., Chan, L., Hiti, A., Johnson, C., and Overturf, G.
(1992). Polysaccharide encapsulated bacterial infection in sickle cell
anemia: a thirty year epidemiologic experience. American Journal of Hematology
39, 176-82.
-
Wright, S., Norris, R., and Mitchell, T. (1992). Ketorolac for sickle cell
vaso-occlusive crisis pain in the emergency department: lack of a
narcotic-sparing
effect. Ann Emerg Med 21, 925-8.
-
Yang, Y., Donnell, C., Farrer, J., and Mankad, V. (1990). Corporectomy
for intractable sickle-associated priapism. Am J Med Sci 300, 231-3.
-
Yaster, M., Tobin, J., Billett, C., Casella, J., and Dover, G. (1994).
Epidural analgesia in the management of severe vaso-occlusive sickle cell
crisis. Pediatrics 93, 310-5.





