Address correspondence to:
Carlo Brugnara, M.D.,
Department of Laboratory Medicine
The Children's Hospital
300 Longwood Avenue, Bader 760
Boston MA 02115
Phone: (617) 355-6610
FAX : (617) 355-6081
e-mail:

Cells with a markedly increased Hb S concentration are a prominent feature
of sickle cell disease, as a consequence of the loss of K, Cl and water
from the erythrocyte. The extreme dependence of polymerization kinetics
on Hb S concentration means that these dehydrated erythrocytes rapidly
sickle when deoxygenated. Blockade of K loss from the erythrocyte should,
therefore, prevent the increase in Hb S concentration and reduce erythrocyte
sickling. Detailed knowledge of the mechanisms leading to cell dehydration
makes this a viable therapeutic option. Two ion transport pathways, the
K-Cl cotransport and the Ca2+-activated
K+
channel play prominent roles in the
dehydration of sickle erythrocytes. Possible therapeutic strategies include
inhibition of K-Cl cotransport by increasing red cell Mg2+content
and inhibition of the Ca2+-activated K
channel by oral administration of clotrimazole.
| AA - normal subjects with red cells containing Hb A; ChTX - Charybdotoxin; DIDS - di-isothiocyano-disulfonyl stilbene; DIOA- [(dihydroindenyl)oxy]alkanoic acid; DTT- dithiothreitol; ISC- irreversibly sickled cells; KTX - kaliotoxin; L1- 1,2-dimethyl-3- hydroxypyrid-4-one; MCHC- mean corpuscular hemoglobin concentration; MCV- mean corpuscular volume; SS - homozygous sickle cell anemia. |
| Note: Parts of this review were taken from: Brugnara, C. Red cell dehydration in pathophysiology and treatment of sickle cell disease. Curr. Op. Hematol. 1995. 2:132-138. |
Hyponatremia have been shown to lead to decreased red cell mean corpuscular hemoglobin concentration (MCHC) and sickling but a subsequent study failed to replicate these findings , suggesting that the levels of clinical hyponatremia required to induce significant cell swelling are difficult to maintain. Additional strategies aimed at inhibiting Hb S polymerization by increasing Hb F concentration have included treatment with hydroxyurea (alone or with erythropoietin) and with butyrate derivatives.
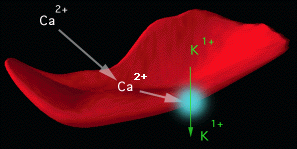
A) Ca2+-activated K+
channel (Gardos pathway):
When the intracellular free Ca2+ concentration
is increased in human red cells, a large K+ loss
with accompanying movement of Cl- and water
is observed. This effect is due to activation of a Ca2+activated
K+
channel, first described by Gardos .
The Gardos channel of human red cells belongs to a family of Ca2+activated
K+channels, present in several cell types
. Ligand binding studies with 125I- charybdotoxin
(ChTX), a peptide toxin, specific inhibitor of the channel, have indicated
the presence of 100-150 binding sites per erythrocyte. The red cell Gardos
channel is inhibited by clotrimazole and other imidazole antimycotics.
|
| Figure 1. Schematic Representation of Gardos Channel Activity in Sickle Erythrocytes. All red cells contain Gardos channel proteins in their membranes. Sickle cells accumulate excessive quantities of calcium, that activate the Gardos channel (illustrated as the blue circle on the red cell membrane.) The Gardos channel is a protein "pump" that expels intracellular potassium when activated by calcium. The red cells loose water along with calcium. The result is a higher intracellular hemoglobin concentration. This promotes polymerization of deoxygenated HbS. |
The cDNA of a high conductance Ca2+-activated K+ channel (maxi-K+ channel) of mouse brain and skeletal muscle has recently been cloned. The molecular identity of the intermediate conductance Gardos channel of red cells remains unknown.
In normal red cells, loss of cell K+ via the Gardos channel plays a role in preventing colloido-osmotic lysis during transient complement activation on the red cell surface . The Gardos channel of sickle cells, either alone or in conjunction with K-Cl cotransport, plays a major role in cell dehydration. In vitro dehydration of sickle erythrocytes depends on external Ca2+ and can be prevented by inhibitors of the Gardos channel such as charybdotoxin, nifedipine or clotrimazole.
B) K-Cl cotransport:
In human red cells, this system was first identified in erythrocytes
of patients homozygous for Hb C disease and subsequently in the least dense,
reticulocyte-rich fraction of normal erythrocytes (AA cells) and in sickle
cells (SS cells). Cell age appears to be one determinant of K-Cl cotransport
activity in human red cells, since the system is active almost exclusively
in normal reticulocytes and not in normal mature red cells. Another important
determinant of this system's activity is the presence of positively charged
hemoglobins (C and S). Studies in different Hb variants have indicated
that activation of K-Cl cotransport is not a common feature of all relatively
positively charged Hb, but is limited to positively charged variants at
ß-6 and ß-7. Activation of K-Cl cotransport is also observed
in ß-thalassemia intermedia erythrocytes. A large portion of this
activation is most likely a consequence of the oxidative damage of the
red cell membrane, since it can be reduced in vitro by exposure to dithiothreitol
(DTT). Studies have shown that SS cells can become denser via the loss
of K+
mediated by the K-Cl cotransport
system. Cytoplasmic acidification of SS erythrocytes leads to activation
of the K-Cl cotransport and dehydration. Cells with a low content of Hb
F display elevated K-Cl cotransport activity, which can account for the
rapid dehydration of reticulocytes and their increase in cell density.
Stimulation of K-Cl cotransport by cell swelling in rabbit and human erythrocytes
is inhibited by protein phosphatases inhibitors.
There are no known inhibitors of sufficient specificity to be used in patients to prevent dehydration via the K-Cl cotransport system. [(Dihydroindenyl)oxy]alkanoic acid, (DIOA), partially inhibits K+ movement via K-Cl cotransport. Red cell Mg2+ content is an important modulator of the activity of this system. Increasing cell Mg2+above the physiologic levels markedly decreases K-Cl cotransport activity. The increase in free cell magnesium levels during deoxygenation is responsible for reducing the volume sensitive flux via K-Cl cotransport in both SS and AA cells. Thus, any maneuver leading to an increase cell Mg2+ content should inhibit dehydration via the K-Cl cotransport system.
Two additional pathways are potentially involved in sickle cell dehydration but their contribution accounts for only a small fraction of the total K+ loss compared with the effect of the two pathways described above. They are:
C) Deoxygenation induced Na and K fluxes and Na-K pump
Original studies by Tosteson and subsequent studies by others have
characterized the increased Na+ and K+
permeabilities
associated with erythrocyte sickling. These deoxygenation induced Na+
and
K+
fluxes are distinguished by their insensitivity
to inhibitors of other red cell transport systems, by their chloride- and
membrane potential-independence and by the inhibitory effect of di-isothiocyano-disulfonyl
stilbene (DIDS). The increased Na+
and
K+ permeability of sickling is associated
with increased entry of Ca2+, which is
also sensitive to DIDS and inhibitors of Ca2+
channels
such as nifedipine. Activation of the Na-K pump, which has a 3 Na+out/2
K+
in stoichiometry, may lead to cell dehydration. Until
recently, no inhibitors of possible clinical used were available for the
deoxygenation fluxes. However, there is evidence for a specific inhibition
of the deoxygenation-induced fluxes by dipyridamole (Persantine¨),
with an effective half-maximal inhibition around 0.5-1 µM. Based
on the known effects of dipyridamole on platelet and endothelial cell functions,
there are now multiple rationales for testing the effect of this drug in
sickle cell disease.
D) Oxidative damage of the cell membrane and K loss
Oxidative damage to the sickle cell membrane is the result of :
 |
Stuart et al. combined CLT with a compound which stabilizes the oxyconformation of Hb S: CLT induced an additive reduction in the rate at which sickle cells clog micropore filters, and may therefore attenuate formation of irreversibly sickled cells.
Studies in a transgenic mouse model for sickle cell disease (SAD mouse) have indicated that CLT inhibits transport and dehydration via the Gardos channel in vitro. Oral administration of CLT (one week at 160 mg/Kg body weight/day or 4 weeks at 80 mg/Kg body weight/day) is associated with marked inhibition of K+ transport via the red cell Gardos channel, increased red cell K+ content, decreased MCHC and density of the red cells. These results in an animal model of sickle cell disease confirm the feasibility of this therapeutic approach of preventing sickle cell dehydration by specifically blocking the Gardos channel.
Studies with normal volunteers taking CLT orally have identified a dosage range (10-20 mg/Kg body weight) which leads to marked inhibition of transport via the red cell Gardos channel . This dosage is substantially lower than that used in the past to treat systemic mycotic infections (60-160 mg/Kg body weight/day). No significant side effects were observed in normal volunteers taking oral CLT for up to 6 days.
We treated 5 subjects who have sickle cell anemia with oral clotrimazole, a specific Gardos channel inhibitor. Patients were started on a dose of 10 mg clotrimazole/kg/day for one week. Protocol design allowed the daily dose to be escalated by 10mg/kg each week until significant changes in erythrocyte density and K+ transport were achieved. Blood was sampled three times a week for hematological and chemical assays, erythrocyte density, cation content and K+ transport. At dosages of 20 mg clotrimazole/kg/day, all subjects showed Gardos channel inhibition, reduced erythrocyte dehydration, increased cell K+ content, and somewhat increased hemoglobin levels. Adverse effects were limited to mild/moderate dysuria in all subjects, and a reversible increase in plasma alanine transaminase and aspartic transaminase levels in two subjects treated with 30 mg clotrimazole/kg/day. This is the first in vivo evidence that the Gardos channel causes dehydration of sickle erythrocytes, and that its pharmacological inhibition provides a realistic anti-sickling strategy. (abstract from Brugnara C, B Gee, C Armsby, S Kurth, M Sakamoto, N Rifai, SL Alper, O. Platt. Therapy with oral clotrimazole induces inhibition of the Gardos channel and reduction of erythrocyte dehydration in patients with sickle cell disease. J. Clin. Invest. 1996; 97: 1227-1234.)
There is a substantial amount of information available on the mechanisms controlling red cell Mg 2+ content. Cell Mg2+ can be either free in the cytoplasm or bound to ATP and 2,3-DPG . The free concentration of Mg2+ is determined not only by the ratio free/bound, but also by the balance between the inward movement of Mg2+ down an electrochemical gradient and the outward movement of Mg2+ via the Mg/Na exchange system. This Mg2+ transport system has been described in detail in chicken and human erythrocytes.
Epidemiological studies have indicated that red cell magnesium content is genetically controlled in normal males, with lower levels in HLA-B35 carriers . It is intriguing to postulate a connection to the observation that certain complications of sickle cell disease are more frequent among patients with HLA-B35. One study reported lower levels of red blood cell magnesium in SS individuals compared with AA individuals. A detailed study on Mg2+ content and transport in sickle cells has indicated that dense SS cells have an abnormally low total Mg2+ content, with a reduced buffering capacity for Mg2+ which leads to an increase of cell free Mg2+. Deoxygenation of SS cells induces a net loss of Mg2+ , because of the increased membrane permeability to Mg2+ and the increased free cell Mg2+ of the dense cells.
Several studies have shown that oral magnesium supplementation can successfully increase erythrocyte magnesium levels. There have been some uncontrolled reports of a beneficial effect of Mg2+ in patients with SS disease. A 7 day course of Mg2+ supplementation did not show any change in red cell survival in 3 patients with SS disease. In transgenic SAD-1 (EMBO J. 1991; 10:3157) and control C576BL/6 mice we investigated the effect of two weeks of diet with either low Mg (6±2 mg/kg body weight/day; n=6) or high Mg (1,000 ± 20 mg/kg body weight/day; n=4), in comparison with a standard diet (Mg: 400 ± 20 mg/kg body weight/day; n=6). High Mg diet increased SAD 1 erythrocyte Mg and K contents, and reduced K-Cl cotransport activity, MCHC, cell density and reticulocyte count. SAD 1 mice treated with low Mg diet showed a significant reduction in erythrocyte Mg and K contents, and increases in K-Cl cotransport, MCHC, cell density, and reticulocyte counts. In SAD mice, Hct and Hb decreased significantly with low Mg diet and increased significantly with high Mg diet. C576BL/6 controls showed significant changes only in erythrocyte Mg and K content, and K-Cl cotransport activities, similar to those observed in SAD mice. Thus, in the SAD mouse, changes in dietary Mg modulate K-Cl cotransport, modify erythrocyte dehydration and ultimately affect Hb levels (from De Franceschi, L., C. Brugnara, and Y. Beuzard. Magnesium intake affects K-Cl cotransport and cell dehydration in transgenic SAD mice: a model for sickle cell disease therapy. Blood 86:299a, 1995).
Due to the low toxicity and side effects of Mg2+ supplementation, this therapeutic approach should be examined carefully for SS disease. Several promising therapeutic strategies based on inhibition of sickle cell dehydration are now at the clinical evaluation stage. If these studies are successful, it seems likely that future therapies of sickle cell disease will be based on combinations of drugs promoting Hb F synthesis, and inhibiting sickle cell dehydration.



