revised May 19, 2000

The Interaction of Iron and Erythropoietin
 The production of red cells involves the coordinate interaction of two organ systems in the body. The first is the bone marrow which produces red cells. The second is the kidney which produces the hormone erythropoietin. Hypoxia (low oxygen) of the kidney prompts synthesis and release of erythropoietin. The erythropoietin then travels to the bone marrow via the blood circulation where it activates new red blood production. This increases the blood's oxygen carrying capacity and corrects the hypoxia which was the primary stimulus to erythropoietin production (Figure 1). Erythropoietin therefore is part of a finely-tuned feedback circuit that controls red blood cell levels. The best analogy is a thermostat and a furnace. The thermostat senses the temperature and the furnace produces heat to eliminate the chill. Once the proper temperature is reached, the thermostat signals the furnace to cease heat production.
The production of red cells involves the coordinate interaction of two organ systems in the body. The first is the bone marrow which produces red cells. The second is the kidney which produces the hormone erythropoietin. Hypoxia (low oxygen) of the kidney prompts synthesis and release of erythropoietin. The erythropoietin then travels to the bone marrow via the blood circulation where it activates new red blood production. This increases the blood's oxygen carrying capacity and corrects the hypoxia which was the primary stimulus to erythropoietin production (Figure 1). Erythropoietin therefore is part of a finely-tuned feedback circuit that controls red blood cell levels. The best analogy is a thermostat and a furnace. The thermostat senses the temperature and the furnace produces heat to eliminate the chill. Once the proper temperature is reached, the thermostat signals the furnace to cease heat production.
Erythropoietin has been cloned and over the past few years studied in great detail. The hormone is a 165 amino acid polypeptide chain. Erythropoietin is heavily glycosylated. That is, the amino acid backbone has a large number of attached sugar molecules (termed complex carbohydrate chains). The protein has a molecular weight of 18 kDa. With the addition of the associated sugars, the apparent molecular weight is 34 kDa. The sugars increase the molecule's stability in the circulation, but not its metabolic activity (Delorme et al., 1992). The carbohydrate chains on the commercial cloned erythropoietin differs from the natural product. The commercially available forms of erythropoietin differ one from the other in patterns of glycocylation. The differences do not affect biological activity (Storring, et al., 1998).
Developing erythroid precursor cells in the bone marrow express erythropoietin receptors at about the BFU-E stage of maturation. The cells each express, at most, 3-400 erythropoietin receptors. In response to erythropoietin and a number of other erythropoietic stimulatory hormones, the red cell precursors produce mature erythrocytes (Figure 2) (Adamson, 1994).
 The distribution of body iron stores shows the importance of iron to red cell production. Normally, about 70% of iron is found in the circulating erythrocytes. Approximately 20% of iron is stored as ferritin, primarily in the liver. Smaller amounts of iron are coupled with enzymes, myoglobin and other proteins. The high iron content of erythrocytes reflects the fact that iron is an integral part of hemoglobin. Hemoglobin comprises over 95% of the protein in red cells. Low grade bleeding (for instance, due to hookworm infection) is the most common cause of iron deficiency worldwide. No physiological means of iron excretion exists. Iron absorption from the gastrointestinal tract is the sole means of regulating iron stores.
The distribution of body iron stores shows the importance of iron to red cell production. Normally, about 70% of iron is found in the circulating erythrocytes. Approximately 20% of iron is stored as ferritin, primarily in the liver. Smaller amounts of iron are coupled with enzymes, myoglobin and other proteins. The high iron content of erythrocytes reflects the fact that iron is an integral part of hemoglobin. Hemoglobin comprises over 95% of the protein in red cells. Low grade bleeding (for instance, due to hookworm infection) is the most common cause of iron deficiency worldwide. No physiological means of iron excretion exists. Iron absorption from the gastrointestinal tract is the sole means of regulating iron stores.
Iron is an essential part of the diet and is absorbed from the duodenum and proximal jejunem. The iron is rapidly transferred to the carrier protein transferrin which delivers it to all the cells in the body. About 80% of absorbed iron is delivered to the bone marrow. A feedback exists such that iron absorption increases when more red cells are produced by the bone marrow. Ironically, some patients with extremely active marrow red cell production can develop iron overload. This occurs in some forms of thalassemia, for instance.
The body has no means of excreting iron. The obligatory daily iron losses are approximately 1-2 mg. The primary modes of iron loss are epithelial shedding from the GI and GU tracts. Epidermal descremation is another source of iron loss. In females, menstruation accounts for approximately 1 mg of iron loss per day on average.
Erythropoiesis involves the close interaction of iron and erythropoietin. In essence, erythropoietin is the accelerator that drives erythropoiesis. Iron is the fuel for the production of new red blood cells. When the two are coupled, red cell production moves briskly and efficiently. If one component is absent (e.g., iron deficiency) anemia results. Even when both components are available, they must be coordinately delivered to the bone marrow for proper action. For instance, if iron arrives on the scene 6 hours after erythropoietin reaches the bone marrow, red cell production will be suboptimal. The erythropoietin would have spent itself on cells that were unable to respond. Little or none of the erythropoietin would be left when the iron finally arrived.
Evidence of the interplay between iron and erythropoietin has existed for a number of years. For instance, subjects who needed surgery for GI bleeding but refused transfusion because of religious beliefs sometimes developed extremely low hematocrits due to very severe iron deficiency. Replacement of iron by intravenous infusion dramatically increased blood cell production. In some reports, hemoglobins rose from 3 g/dl to 9 g/dl over the course of 14 days (Dudrick, et al,. 1985). These studies were performed before plasma erythropoietin levels could be determined. However, we can extrapolate from current data that the subjects had extremely high erythropoietin levels. The only block to new red cell production was their iron deficiency. When the two components iron and erythropoietin are brought together, bone marrow activity surges tremendously.
On the opposite side of the coin are subjects who have very high iron stores but very low erythropoietin levels. Historically this was seen most clearly in patients with chronic renal failure who required hemodialysis. Because these subjects lacked kidney function, they not only failed to excrete toxins from the body via the kidney, they also failed to produce the erythropoietin needed for erythropoiesis. With the cloning of the erythropoietin gene, the drug became available for use in the late 1980's. The hematocrit of patients with chronic renal failure rose dramatically with erythropoietin treatment (Eschbach, et al., 1987). Iron stores were quite high in these early subject all of whom had been on maintenance transfusion therapy prior to the introduction of erythropoietin. Iron overload from transfusions had been a serious problem. Ironically, one group of investigators phlebotomized dialysis patients to reduce iron stores following the introduction of erythropoietin for clinical use (Lazarus, et al., 1990).
Erythropoietin increased both the red cell mass and hematocrit in these subjects. Enhanced cell production manifested itself as a rise in the plasma iron turnover (PIT) and a decrease in the half-time of iron in the plasma. Interestingly, the plasma ferritin level of patients dropped substantially despite quite adequate iron stores. This was the first indication that erythropoietin used in high doses could drive red cell production more rapidly than iron could be delivered to the bone marrow even in subjects with adequate iron stores (Hotta, et al. 1991).
A study by Rutherford and colleagues in 1994 dramatically demonstrated the interplay between iron and erythropoietin (Rutherford, et al. 1994). Normal male volunteers were placed on oral iron supplementation in a study to determine whether erythropoietin could raise the baseline hemoglobin to an extent that enhanced autologous blood donation was possible. The study included only males to reduce the possibility of underlying iron deficiency which occurs commonly in normal female subjects. The addition of oral iron during the erythropoietin treatment was believed to provide an adequate source of iron for the production new red blood cells. The subjects received one of three dosing regimens of erythropoietin over 14 days and then were followed-up at day 24. In all cases, the baseline hemoglobin increased in these subjects by approximately 1g/dl. This increase reflected the enhanced erythropoiesis produced by erythropoietin supplementation. The elevated hemoglobin level persisted for as long as 24 days after the initial infusion.
Surprisingly, the subjects were unable to maintain adequate serum iron levels despite the oral iron supplementation. Transferrin saturation at the onset of the study was approximately 40%, a value which is normal for adult males. After 13 days of erythropoietin treatment, the average transferrin saturation fell to about 10%. This drop showed that developing red cells removed iron from the circulating transferrin at a rate faster than it could be replaced, either from iron stores or from orally absorbed iron. With the cessation of erythropoietin treatment the plasma transferrin saturation returned to its baseline of about 40%. This was because the red cell precursors were no longer pushed into overdrive activity by surplus erythropoietin. Erythropoietin availability and iron use returned to their normal balance.
Even more surprisingwas the fall in plasma ferritin levels from the range of 100 ng/dl to approximately 20 ng/dl. With the cessation of erythropoietin treatment, the plasma ferritin levels began to rise towards their baseline levels. Plasma ferritin is believed to reflect iron stores. Since the increase in hemoglobin was only about 1 g/dl, the two weeks of erythropoietin therapy did not substantially reduce total body iron stores in these patients. These data indicate that the plasma ferritin level may reflect iron stores at steady state. However, when the steady state of erythropoiesis is disturbed, such as with the use of exogenous erythropoietin, the plasma ferritin value no longer correlates with iron stores.
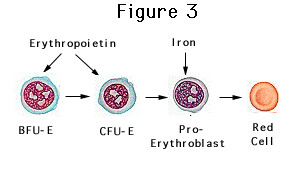 Our model of iron and erythropoietin shows stem cells maturing along the erythroid line to form BFU-E's (Figure 3). At this point iron is needed for the production of new hemoglobin. Erythropoietin promotes the maturation of BFU-E's into CFU-E's and subsequently into mature erythrocytes. If erythropoietin is present without sufficient iron, there is insufficient fuel for red cell production. This is the scenario in subjects with severe iron deficiency. However, impaired erythropoiesis also occurs in patients in whom red cell production is suddenly jolted forward by a burst of erythropoietin from an injection of the hormone. This condition has been termed, functional iron deficiency. Investigators have documented that the transient, supraphysiologic levels of hormone seen with erythropoietin injection produces hypochromic red cells similar to those seen with frank iron deficiency (Madore, et al., 1997).
Our model of iron and erythropoietin shows stem cells maturing along the erythroid line to form BFU-E's (Figure 3). At this point iron is needed for the production of new hemoglobin. Erythropoietin promotes the maturation of BFU-E's into CFU-E's and subsequently into mature erythrocytes. If erythropoietin is present without sufficient iron, there is insufficient fuel for red cell production. This is the scenario in subjects with severe iron deficiency. However, impaired erythropoiesis also occurs in patients in whom red cell production is suddenly jolted forward by a burst of erythropoietin from an injection of the hormone. This condition has been termed, functional iron deficiency. Investigators have documented that the transient, supraphysiologic levels of hormone seen with erythropoietin injection produces hypochromic red cells similar to those seen with frank iron deficiency (Madore, et al., 1997).
In situations where supraphysiological spurts of erythropoietin force red cell production that outstrips the availability of iron, the patients produce protoporphyrin IX, the precursor the heme, but lack the iron that normally is inserted into the protoporphyrin. Zinc, the second most abundant divalent cation in the red cell, fills the void to form zinc protoprophyrin (ZPP). Unfortunately, zinc protoporphyrin does not function in oxygen delivery by hemoglobin. Red cells normally contain no zinc protophorphyrin due to the exquisite balance in the availability of iron and erythropoietin. A simple, clinically available test detects zinc protoporphyrin because the molecule is fluorescent. Zinc protoprophyrin in the red cells of people who receive intermittent injections of erythropoietin indicates suboptimal hemoglobin production because either the patient is iron deficient or lacks the ability to mobilize iron from stores sufficently quickly to meet the need created by the exogenously administered hormone (Fishbane and Lynn, 1995).
The implications of this model for the clinical use of erythropoietin are great. Erythropoietin is recommended for the treatment of the anemia of chronic renal failure (Eshbach, et al., 1991). The hormone is also used in people with anemia due to AZT and other treatments of AIDS, transfusion dependent myelodysplasia, chemotherapy induced anemia and anemia in surgical settings. Iron deficiency will render any of these patients less responsive to erythropoietin than normally would be the case. In many instances, the baseline responsiveness to erythropoietin is already hampered by the "anemia of chronic inflammation" as well as primary damage to the erythrocyte producing cells (Goldwasser, et al., 1994). Careful evaluation of the subject's iron stores is necessary to utilize erythropoietin most effectively.
Even patients with "normal" iron stores may respond poorly to erythropoietin therapy. The result is dampened red cell production relative to that seen with abundant iron stores. Patients treated with erythropoietin can increase the quantity of banked blood for possible autologous transfusion prior to surgery (Rutherford et al., 1994). Parenteral iron administration along with the erythropoietin avoids functional iron deficiency and the associated limited red cell production. Table 1 provides a schema for administration of iron dextran. An iron saccharate compound (Ferrilecit ®) has been recently released for dialysis patients in the US receiving erythropoietin therapy. The compound is simliar to those used in Europe for 25 years because of their better tolerance and greater safety relative to iron dextran. Use of Ferrilecit® in any other fashion would be off-label.
References
- Adamson, JW. 1994. The relationship of erythropoietin and iron metabolism to red blood cell production in humans. Semin Oncol 21(2 suppl 2): 9-15.
- Delorme E, Lorenzini T, Giffin J, Martin F, Jacobsen F, Boone T, Elliott S. 1992. Role of glycosylation on the secretion and biological activity of erythropoietin. Biochemistry 31:9871-9876.
- Dudrick SJ, O'Donnell JJ, Raleigh DP, Matheny RG, Unkel SP. 1985. Rapid restoration of red blood cell mass in severely anemic surgical patients who refuse transfusion. Arch Surg 120:721-727.
- Eschbach JW, Egrie JC, Dwoning MR, Browne JK, Adamson JW. 1987. Correction of the anemia of end-stage renal disease with recombinant human erythropoietin. N Engl J Med 316:73-78.
- Eschbach JW, Haley NR, Adamson JW. 1990 New insights into the treatment of the anemia of chronic renal failure with erythropoietin. Semin Dial 3:112-121.
- Fishbane S, Lynn RI. 1995. The utility of zinc protoporphyrin for predicting the need for intravenous iron therapy in hemodialysis patients. Am J Kidney Dis 25:426-432
.
- Goldwasser E, Koutelos, T, Abraham S, Avram MM. 1994. Serum ferritin, hematocrit and mean corpuscular volume in hemodialysis. Nephron 67:30-35.
- Hotta T, Ogawa H, Saito, A, Ito A. 1991. Iron balance following recombinant human erythropoietin therapy for anemia associated with chronic renal failure. Int J Hematol 54:195-200.
- Lazarus JM, Hakim RM, Newell J. 1990. Recombinant human erythropoietin and phlebotomy in the treatment of iron overload in chronic hemodialysis patients. Am J Kidney Dis 16:101-108.
- Madore F, Lowrie EG, Brugnara C, Lew NL, Lazarus JM, Bridges K, Owen WF. 1997. Anemia in hemodialysis: variables affecting this outcome predictor. J Am Soc Nephrol :8(12):1921-1929.
- Madura, JA. 1993. Use of erythropoietin and parenteral iron dextran in a severely anemic Jehovah's Witness with colon cancer. Arch Surg 128:1168-1170.
- Rutherford CJ, Schneider TJ, Dempsey H, Kirn DH, Brugnara C, Goldberg MA. 1994. Efficacy of different dosing regimens for recombinant human erythropoietin in a simulated perisurgical setting: the importance of iron availability in optimizing response. Am J Med 96:139-145.
- Storring PL, Tiplady RJ, Gaines Das RE, Stenning BE, Lamikanra A, Rafferty
B, Lee J. 1998. Epoetin alfa and beta differ in their erythropoietin isoform
compositions and biological properties. Br J Haematol 100:79-89




