
The average adult stores about 1 to 3 grams of iron in his or her body. An exquisite balance between dietary uptake and loss maintains this balance. About 1 mg of iron is lost each day through sloughing of cells from skin and mucosal surfaces, including the lining of the gastrointestinal tract (Cook et al., 1986). Menstration increases the average daily iron loss to about 2 mg per day in premenopausal female adults (Bothwell and Charlton, 1982). No physiologic mechanism of iron excretion exists. Consequently, absorption alone regulates body iron stores (McCance and Widdowson, 1938). The augmentation of body mass during neonatal and childhood growth spurts transiently boosts iron requirements (Gibson et al., 1988).
|
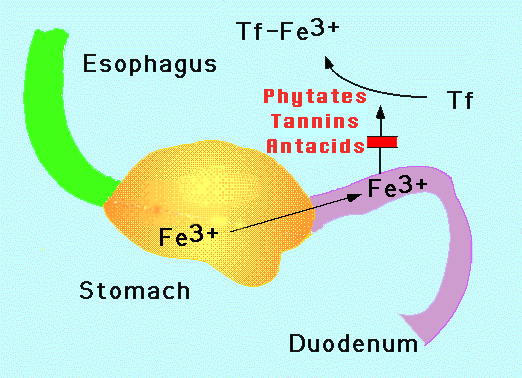
| Figure 1. Iron absorption. Iron enters the stomach from the esophagus. Iron is oxidized to the Fe3+ state no matter its original form when taken in orally. Gastric acidity as well as solubilizing agents such as ascorbate prevent precipitation of the normally insoluble Fe3+ . Intestinal mucosal cells in the duodenum and upper jejunum absorb the iron. The iron is coupled to transferrin (Tf) in the circulation which delivers it to the cells of the body. Phytates, tannins and antacids block iron absorption. |
The physical state of iron entering the duodenum greatly influences its absorption however. At physiological pH, ferrous iron (Fe2+) is rapidly oxidized to the insoluble ferric (Fe3+) form. Gastric acid lowers the pH in the proximal duodenum, enhancing the solubility and uptake of ferric iron (Table 1). When gastric acid production is impaired (for instance by acid pump inhibitors such as the drug, prilosec), iron absorption is reduced substantially.
Heme is absorbed by machinery completely different to that of inorganic
iron. The process is more efficient and is independent of duodenal pH .
Consequently meats are excellent nutrient sources of iron. In fact, blockade
of heme catabolism in the intestine by a heme oxygenase inhibitor can produce
iron deficiency (Kappas et al., 1993). The paucity of meats in the diets
of many of the people in the world adds to the burden of iron deficiency.
A number of dietary factors influence iron absorption. Ascorbate and citrate increase iron uptake in part by acting as weak chelators to help to solubilize the metal in the duodenum (Table 1) (Conrad and Umbreit, 1993). Iron is readily transferred from these compounds into the mucosal lining cells. Conversely, iron absorption is inhibited by plant phytates and tannins. These compounds also chelate iron, but prevent its uptake by the absorption machinery (see below). Phytates are prominent in wheat and some other cereals, while tannins are prevalent in (non-herbal) teas.
Lead is a particularly pernicious element to iron metabolism (Goya, 1993). Lead is taken up by the iron absorption machinery, and secondarily blocks iron through competitive inhibition. Further, lead interferes with a number of important iron-dependent metabolic steps such as heme biosynthesis. This multifacted attack has particularly dire consequences in children, were lead not only produces anemia, but can impair cognitive development. Lead exists naturally at high levels in ground water and soil in some regions, and can clandestinely attack children's health. For this reason, most pediatricians in the U.S. routinely test for lead at an early age through a simple blood test.
Immaturity of the gastrointestinal tract can exacerbate iron deficiency
in newborns. The gastrointestinal tract does not achieve competency for
iron absorption for several weeks after birth. The problem is even more
severe for premature infants, who tend to be anemic for a variety of reasons.
A substantial portion of iron stores in newborns are transferred from the
mother late in pregnancy. Prematurity shortcircuits this process. Parenteral
iron replacement is possible, but not often used because of the often delicate
health of premature infants. Transfusion becomes the default option in
this circumstance.
| Physical State (bioavailability) | heme > Fe2+ > Fe3+ |
| Inhibitors | phytates, tannins, soil clay, laundry starch, iron overload, antacids |
| Competitors | lead, cobalt, strontium, manganese, zinc |
| Facilitators | ascorbate, citrate, amino acids, iron deficiency |
The mechanism by which iron enters the mucosal cells lining the upper
gastrointestinal tract is unknown. Most cells in the rest of the body are
believed to acquire iron from plasma transferrin (an iron-protein chelate),
via specific transferrin receptors and receptor-mediated endocytosis (Klausner,
et al, 1983). The hypothesis that apotransferrin (or an equivalent molecule)
secreted by intestinal cells or present in bile chelates intestinal iron
and facilitates its absorption(Huebers et al., 1983) is unsubstantiated.
The transferrin gene is not expressed in intestinal cells. Later work indicated
that transferrin found in the intestinal lumen is derived from plasma (Idzerda
et al., 1986). Plasma transferrin entering bile is fully saturated with
iron, obviating any intraluminal chelating function (Schumann et al., 1986).
Furthermore, hypoxia, which greatly increases iron absorption, has no effect
on intestinal transferrin levels (Simpson et al., 1986). Exogenous transferrin
cannot donate iron to intestinal mucosal cells (Bezwoda et al., 1986),
and the brush boarder membrance lacks transferrin receptors (Parmley et
al., 1985) (although they are present on the basolateral surface of intestinal
epithelial cells (Levin et al., 1984); (Banerjee et al., 1986). Lastly
and perhaps most compellingly, humans and mice with hypotransferrinemia
paradoxically absorb more dietary iron than normal. Although the erythron
is iron deficient, these individuals develop hepatic iron overload (Heilmeyer
et al., 1961); (Craven et al., 1987).
Ý
A very different scheme of iron uptake has been proposed by investigators
studying iron transport in yeast. Yeast face the problem of taking in iron
from the environment, a process similar to that of intestinal mucosal cells.
Dancis et al. used genetic selection to isolate Sacchromyces cerevisiae
mutants with defective iron transport (Dancis et al., 1994); (Stearman
et al., 1996). They constructed an expression plasmid in which an enzyme
necessary for histidine biosynthesis was under the control of an iron-repressible
promoter. The plasmid was introduced into a yeast histidine auxotroph (i.e.
a strain of yeast that requires histidine to survive). Mutants were selected
in the absence of histidine, in the presence of high levels of iron. Among
the mutats they isolated, were cells with defective iron uptake. They discovered
that membrane iron transport depends absolutely upon copper transport.
In this model, ferric iron in yeast culture medium is reduced to its ferrous
form by an externally oriented reductase (FRE1). The element is shuttled
rapidly into the cell by a ferrous transporter, which appears to be coupled
to an externally oriented copper-dependent oxidase (FET3) embedded in the
cell membrane (De Silva et al., 1995); (Stearman et al., 1996). FET3 is
strikingly homologous to the mammalian copper oxidase ceruloplasmin. The
re-oxidation of ferrous to ferric iron is apparently an obligatory step
in the transport mechanism, although the coupling mechanism of oxidation
and membrane transport is unclear. (De Silva et al., 1995); (Stearman et
al., 1996); (Yuan et al., 1995). Although the genetic evidence for this
scheme is compelling, the central component, the ferrous transporter itself,
remains elusive. These investigators speculate that mammalian intestinal
iron transport is analogous to the yeast iron uptake process (Harford et
al., 1994). This assertion is supported by studies of copper-deficient
swine, which show co-existing iron deficiency unresponsive to iron therapy
(Lahey et al., 1952); (Gubler et al., 1952); (Cartwright et al., 1956).
Ý
ÝMice that are homozygous or heterozygous for the sla mutation (sla/sla or sla/y) also have low serum iron levels. In contrast to mk mice, they have abnormal iron deposits within intestinal mucosal cells, suggesting that this X-linked defect impairs intracellular iron trafficking or basolateral export of iron to the plasma. The sla animals differ further from the mk mice by correction of anemia by parenteral iron. Based on studies of these mutants, distinct apical and basolateral iron transport systems possibly exist that function coordinately to transfer iron from intestinal lumen to plasma.
ÝWhatever the mechanism of iron uptake, normally only about 10% of the elemental iron entering the duodenum is absorbed. However, this value increases markedly with iron deficiency (Finch, 1994). In contrast, iron overload reduces but does not eliminate absorption, reaffirming the fact that absorption is regulated by body iron stores. In addition, both anemia and hypoxia boost iron absorption. A portion of the iron that enters the mucosal cells is retained sequestered within ferritin. Intracellular intestinal iron is lost when epithelial cells are sloughed from the lining of the gastrointestinal tract. The remaining iron traverses the mucosal cells, to be coupled to transferrin for transport through the circulation.
ÝA circulating factor related to erythropoiesis that modulates iron absorption has been hypothesized, but not identified (Beutler and Buttenweiser, 1960); (Finch, 1994). Several candidate factors have been excluded, including transferrin (Aron et al., 1985) and erythropoietin (Raja et al., 1986). Clinical manifestations of this apparent communication between the marrow and the intestine includes iron overload that develops in patients with severe thalassemia in the absence of transfusion. The accelerated (but ineffective) erythropoiesis in this condition substantially boosts iron absorption. In some cases, the coupling of increased PIT and increased gastrointestinal iron absorption is beneficial. In pregnancy, placental removal of iron raises the PIT. This process enhances gastrointestinal iron absorption thereby increasing the availability of the element to meet the needs of the growing and developing fetus.
ÝCompetition studies suggest that several other heavy metals share the iron intestinal absorption pathway. These include lead, manganese, cobalt and zinc (Table 1). Enhanced iron absorption induced by iron deficiency also augments the uptake of these elements. As iron deficiency often coexists with lead intoxication, this interaction can produce particularly serious medical complications in children (Piomelli et al., 1987). Interestingly, copper absorption and metabolism appear to be handled mechanisms different to those of iron.
ÝÝÝ




