
|
 |
| Figure 1. Inheritance of sickle genes from parents with sickle cell trait. As shown in the graphic, the couple has one chance in four that the child will be normal, one chance in four that the child will have sickle cell disease, and one chance in two that the child will have sickle cell trait. |
A person receives the sickle cell genes or not only at the time of conception. Therefore, neither sickle cell trait nor sickle cell disease can be contracted. By the same token, people cannot lose their sickle cell genes over time. A person born with sickle cell trait (one sickle cell gene) will always have sickle cell trait. The same is true of sickle cell disease (two sickle cell genes). Sickle cell disease produces illness, while sickle cell trait usually does not. The severity of sickle cell disease can change over time. The change in severity is not due to a change in the sickle cell genes over time. Rather, number of other biological factors, most of which are not understood, change to alter the severity of sickle cell disease.
People who inherit two genes for sickle hemoglobin (one from each parent) have sickle cell disease. With a few exceptions, a child can inherit sickle cell disease only if both parents have one gene for sickle cell hemoglobin. The most common situation in which this occurs is when each parent has one sickle cell gene. In other words, each parent has sickle cell trait. Figure 1 shows the possible combination of genes that can occur for parents each of whom has sickle cell trait.
A one-in-four chance exists that a child will inherit two normal genes from the parents. A one-in-four chance also exists that a child will inherit two sickle cell genes, and have sickle cell disease. A one-in-two chance exists that the child will inherit a normal gene from one parent and a sickle gene from the other. This would produce sickle trait.
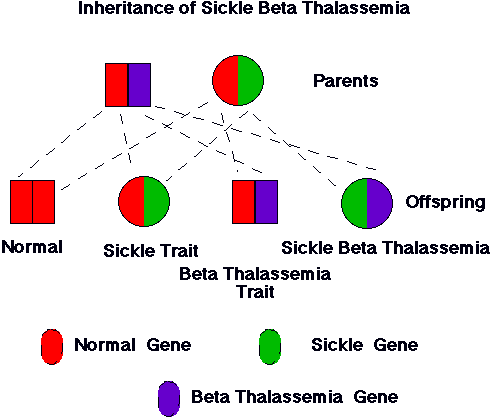 |
| Figure 2. Inheritance of hemoglobin genes from parents with sickle cell trait and thalassemia trait. As illustrated, the couple has one chance in four that the child will have the genes both for sickle hemoglobin and for thalassemia. The child would have sickle b-thalassemia. The severity of this condition is quite variable. The nature of the thalassemia gene (bo or b+) greatly influences the clinical course of the condition. |
These probabilities exist for each child independently of what happened with prior children the couple may have had. In other words, each new child has a one-in-four chance of having sickle cell disease. A couple with sickle cell trait can have eight children, none of whom have two sickle genes. Another couple with sickle trait can have two children each with sickle cell disease. The inheritance of sickle cell genes is purely a matter of chance. These probability odds cannot be altered.
Another hemoglobin disorder that interacts with sickle cell disease is hemoglobin C. The abnormal hemoglobin C protein is relatively harmless. People with two hemoglobin C genes have a relatively mild clinical condition termed "hemoglobin C disease". When hemoglobin C combines with hemoglobin S, the result is "hemoglobin SC disease". On average, hemoglobin SC disease is milder than sickle cell disease. However, some patients with hemoglobin SC disease have a clinical condition as severe as any with sickle cell disease. The reason for the marked variability in the clinical course of hemoglobin SC disease is unknown. We do know that the tendency of hemoglobin C to produce red cell dehydration is a major reason that the combination of hemoglobins S and C is so problematic.
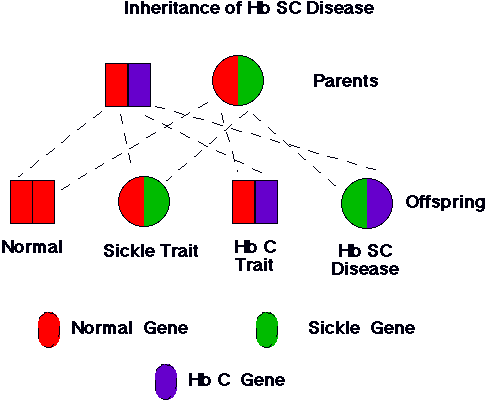 |
| Figure 3. Inheritance of hemoglobin genes from parents with sickle cell trait and hemglobin C trait. As illustrated, the couple has one chance in four that the child will have the genes both for sickle hemoglobin and for hemoglobin C. The child would have hemoglobin SC disease. Hemoglobin SC disease is generally milder than sickle cell disease. Unfortunately, some patients run a clinical course that is indistiguishable from sickle cell disease. |
Newborns in most states are tested at birth by hemoglobin electrophoresis that detects sickle cell disease, sickle cell trait and hemoglobin C. For technical reasons, newborn screening by electrophoresis often does not detect b-thalassemia trait. Newborn screening assures that children with sickle cell disease will receive proper care. In the past, some children died of complications of their sickle cell disease before the condition was diagnosed.
A problem exists with children who come to the US as immigrants. Rarely have these children been screened at birth for sickle cell disease. If the diagnosis has not been made on the basis of symptoms, these children can slip through the treatment net and fall victim to preventable complications of sickle cell disease. In particular, children between the ages of 6 months and 7 years should receive penicillin (or erythromycin if they have a penicillin allergy) to reduce the chance of overwhelming and frequently fatal bacterial infections. For this reason, some authorities in pediatric sickle cell disease advocate screening children from high risk groups whenever they enter a pediatric practice.
Children who move between states can also present a problem with respect to the diagnosis of sickle cell disease. Parents sometimes forget the results of newborn screening, particularly if a child has relatively mild sickle cell disease. Unfortunately, children with otherwise mild sickle cell disease are at equal risk of death from overwhelming bacterial infection. No national registry exists for children with sickle cell disease. In the absence of such a data base, tracking down the results of screening tests performed in other states (or even the same state) can be a formidable task. When weighed against the chance of a preventable death, even an "extra" screen for sickle cell disease for a child who may have been tested in another state is a good investment.




