
The inheritance pattern is complicated in patients with thalassemia because two sets of genes on different chromosomes cooperate to produce hemoglobin. A defect anywhere in this complex can produce thalassemia. The expression of thalassemia, therefore, more closely resembles that of height, with gradations in effect.
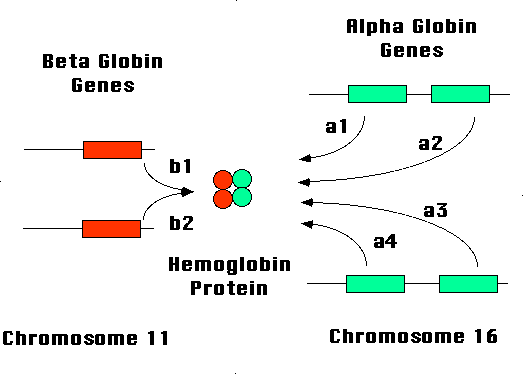
Each hemoglobin molecule contains four subunit proteins. Two of the subunit proteins are called alpha and two are called beta. Hemoglobin properly binds and releases oxygen only when two alpha subunits are connected to two beta subunits. A pair of genes located on chromosome #16 controls the production of the alpha subunits of hemoglobin. A single gene located on chromosome #11 controls the production of the hemoglobin beta subunit (Figure 1).
All cells contain pairs of idential chromosomes, one from the father and one from the mother. Each chromosome contains thousands of genes lined up in sequence. As shown in Figure 1, each person has two beta globin genes, one from the father and one from the mother. Since each chromosome #16 has two alpha globin genes, each person has a four of these genes. One chromosome #16 comes from the father, who therefore contributes two alpha globin genes to the offspring. One chromosome #16 comes from the mother who also contributes two alpha globin genes to the offspring.
A complete hemoglobin molecule has four subunits: two alpha and two beta. The two beta globin genes contribute equally to the production of beta globin subunit protein. The alpha globin genes together produce an amount of alpha globin protein that exactly equals the beta globin protein. Since there are four alpha globin genes compared to two beta globin genes, each alpha globin gene produces only about half as much protein as a beta globin gene. These keeps the overall production of subunits equal from each set of chromosomes (Figure 1).
Thalassemia occurs when one or more of the genes fails to produce protein, leading to a shortage of one of the subunits. If one of the beta globin genes fails, the condition is called beta thalassemia. Beta thalassemia, therefore, is due to a shortage of beta subunits. If an alpha globin gene fails, the condition is called alpha thalassemia. In this case, a shortage of alpha subunits occurs.
| Figure 1. The two chromosomes #11 have one beta globin gene each (for a total of two genes). The two chromsomes #16 have two alpha globin genes each (for a total of four genes). Hemoglobin protein has two alpha subunits and two beta subunits. Each alpha globin gene produces only about half the quantity of protein of a single beta globin gene. This keeps the production of protein subunits equal. Thalassemia occurs when a globin gene fails, and the production of globin protein subunits is thrown out of balance. |
The beta globin genes exist in the cell, but fail to operate normally in beta thalassemia. In some cases, the gene failure is not total. The gene produces a small amount of normal beta protein. Sometimes, a person inherits two beta thalassemia genes in which the production of beta globin protein from each is reduced, but is not zero. The resulting clinical condition is more severe that thalassemia minor, where one gene fails but the other works normally. The condition is less severe than thalassemia major, where both beta globin genes fail completely. The clinical condition is termed, thalassemia intermedia.
A variety of different beta thalassemia genes cause only a partial failure in beta globin protein production. In their partial functioning, some of these genes produce more beta globin protin than others. For this reason, the course of thalassemia intermedia varies greatly between patients. A person with two defective beta globin genes that nonetheless produce a reasonable amount of beta globin protein can have a clinical condition that is close to someone with thalassemia minor. In other instances, the two failing genes produce very little beta globin protein, and the person has a clinical condition similar to thalassemia major. Thalassemia intermedia is a clinical condition that varies and must be constantly evaluated by the hematologist. No two people with thalassemia intermedia are the same.
Thalassemia minor (or trait) is usually a benign condition that produces only a mild anemia. The more severe forms of thalassemia occur when a person inherits two thalassemia genes. Figure 2 shows the possible outcomes for offspring of two people with thalassemia minor. A one-in-four chance exists that a child will inherit two normal genes from the parents. A one-in-four chance also exists that a child will inherit two thalassemia genes, and have a severe form of thalassemia (thalassemia major or thalassemia intermedia). A one-in-two chance exists that the child will inherit a normal gene from one parent and a thalassemia gene from the other. This would produce thalassemia minor (or trait).
These probabilities exist for each child independently of what happened with prior children the couple may have had. In other words, each new child has a one-in-four chance of having severe thalassemia. A couple in which each has thalassemia trait can have eight children, none of whom have two thalassemia genes. Another similar couple can have two children each with severe thalassemia. The inheritance of thalassemia genes is purely a matter of chance and cannot be altered.
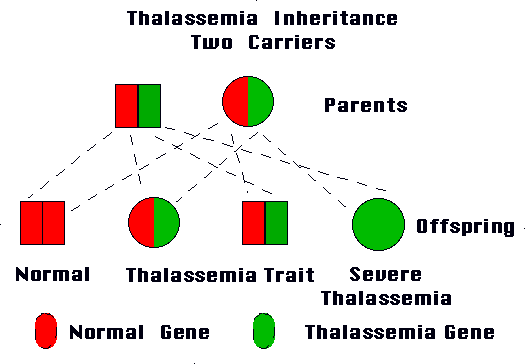 | Figure 2. Inheritance of hemoglobin genes from parents with thalassemia trait. As illustrated, the couple has one chance in four that a child will inherit two thalassemia genes. The child would have a severe form of thalassemia (thalassemia major or thalassemia intermedia). The severity varies, often significantly. The nature of the particular thalassemia genes greatly influences the clinical course of the disorder. |
The clinical severity of the thalassemia in a person who inherits two thalassemia genes will depend on the amount of beta globin protein produced by the defective genes. Some thalassemia genes produce essentially no beta globin protein, and are called beta0 thalassemia genes. A person with two such genes has severe, transfusion-dependent thalassemia, called thalassemia major.
Often, the thalassemia genes produce some beta globin protein, but the amount is reduced. These thalassemia genes are called beta+ thalassemia genes. A person who has one beta+ and one beta0 thalassemia gene will have thalassemia major. Usually, a person with two beta+ thalassemia genes also requires chronic transfusion therapy, and therefore also has thalassemia major.
As noted, some beta+ thalassemia genes produce reduced, but reasonable amounts of beta globin protein. Two such genes can sometimes together produce enough beta globin protein so that the patient does not require chronic transfusions to live. This condition is thalassemia intermedia. Thalassemia intermedia is defined clinically by the transfusion requirement of the patient. Many considerations go into the decision to transfuse a patient chronically. Therefore, a person can change clinically from thalassemia intermedia to thalassemia major at some point during their life, while no chance occurs in their genetic makeup.
The key issue is whether two alpha genes on the same chromosome are deleted. If so, the offspring has the chance of having a very severe alpha thalassemia condition in which two alpha globin genes are missing on one chromosome #16, and one is missing on the other chromsome #16 (Figure 3). In that instance, only the person has only one functional alpha globin gene. The result is a severe, transfusion-dependent anemia called Hemoglobin H Disease. If all four alpha globin genes are missing, the condition is incompatible life. Most fetuses die in utero with this condition (hydrops fetalis). Alpha thalassemia in which two genes are missing on the same chromosome occurs commonly in people of Asian ancestry.
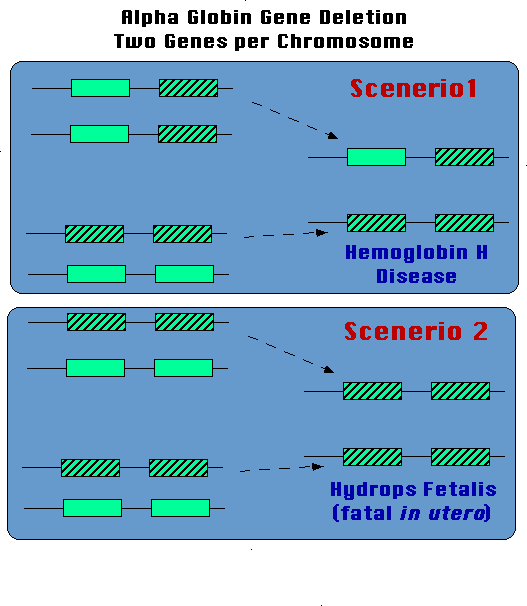 | Figure 3. People of Asian ancestry often have two alpha globin genes deleted on the same chromosome #16. The parents each have the mild thalassemia that results with two functioning alpha globin genes. The offspring that inherits the double deletion from one parent and the single from the other will have Hemoglobin H disease (Scenario 1). The offspring who inherits no alpha genes from the parents dies in utero (Scenario 2; hydrops fetalis). |
Alpha thalassemia also occurs in people of African ancestry. Here, the loss of two alpha globin genes on the same chromosome #16 is extremely rare (Figure 4). Consequently, alpha thalassemia is usually mild in such patients. Hemoglobin H disease and hydrops fetalis are exceedingly uncommon.
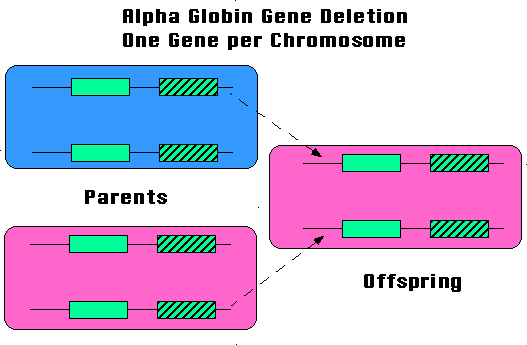 | Figure 4. People of African ancestry usually have only one alpha globin gene deleted per chromosome. The parents each have the mild thalassemia that results with two functioning alpha globin genes. The offspring can, at most, inherit the relatively mild condition of the parents. |



